| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |


Andrzej Zielezinski, Adam Gudyś, Jakub Barylski, Krzysztof Siminski, Piotr Rozwalak, Bas E. Dutilh, and Sebastian Deorowicz (2024) "Ultrafast and accurate sequence alignment and clustering of viral genomes", Nature Methods, doi: 10.1038/s41592-025-02701-7.
Viromics produces millions of viral genomes and fragments annually, overwhelming traditional sequence comparison methods. Here we introduce Vclust, an approach that determines average nucleotide identity by Lempel–Ziv parsing and clusters viral genomes with thresholds endorsed by authoritative viral genomics and taxonomy consortia. Vclust demonstrates superior accuracy and efficiency compared to existing tools, clustering millions of genomes in a few hours on a mid-range workstation.


Tatiana Demina, Heli Marttila, Igor S. Pessi, Minna K. Männistö, Bas E. Dutilh, Simon Roux, and Jenni Hultman (2025) "Tunturi virus isolates and metagenome-assembled viral genomes provide insights into the virome of Acidobacteriota in Arctic tundra soils", Microbiome 13: 79, doi: 10.1186/s40168-025-02053-6, Pubmed. Preprint.
Background Arctic soils are climate-critical areas, where microorganisms play crucial roles in nutrient cycling processes. Acidobacteriota are phylogenetically and physiologically diverse bacteria that are abundant and active in Arctic tundra soils. Still, surprisingly little is known about acidobacterial viruses in general and those residing in the Arctic in particular. Here, we applied both culture-dependent and -independent methods to study the virome of Acidobacteriota in Arctic soils. Results Five virus isolates, Tunturi 1-5, were obtained from Arctic tundra soils, Kilpisjärvi, Finland (69°N), using Tunturiibacter spp. strains originating from the same area as hosts. The new virus isolates have tailed particles with podo- (Tunturi 1, 2, 3), sipho- (Tunturi 4), or myovirus-like (Tunturi 5) morphologies. The dsDNA genomes of the viral isolates are 63-98 kbp long, except Tunturi 5, which is a jumbo phage with a 309-kbp genome. Tunturi 1 and Tunturi 2 share 88% overall nucleotide identity, while the other three are not related to one another. For over half of the open reading frames in Tunturi genomes, no functions could be predicted. To further assess the Acidobacteriota-associated viral diversity in Kilpisjärvi soils, bulk metagenomes from the same soils were explored and a total of 1881 viral operational taxonomic units (vOTUs) were bioinformatically predicted. Almost all vOTUs (98%) were assigned to the class Caudoviricetes. For 125 vOTUs, including five (near-)complete ones, Acidobacteriota hosts were predicted. Acidobacteriota-linked vOTUs were abundant across sites, especially in fens. Terriglobia-associated proviruses were observed in Kilpisjärvi soils, being related to proviruses from distant soils and other biomes. Approximately genus- or higher-level similarities were found between the Tunturi viruses, Kilpisjärvi vOTUs, and other soil vOTUs, suggesting some shared groups of Acidobacteriota viruses across soils. Conclusions This study provides acidobacterial virus isolates as laboratory models for future research and adds insights into the diversity of viral communities associated with Acidobacteriota in tundra soils. Predicted virus-host links and viral gene functions suggest various interactions between viruses and their host microorganisms. Largely unknown sequences in the isolates and metagenome-assembled viral genomes highlight a need for more extensive sampling of Arctic soils to better understand viral functions and contributions to ecosystem-wide cycling processes in the Arctic.
Amruta Nair, Swapnil Prakash Doijad, Mangesh Vasant Suryavanshi, Anwesha Dey, Satya Veer Singh Malik, Bas E. Dutilh, and Sukhadeo Baliram Barbuddhe (2025) "Unveiling the Kadaknath gut microbiome: early growth phase spatiotemporal diversity", Microbiology Research 16: 54, doi: 10.3390/microbiolres16030054 .
The early growth phase is a critical period for the development of the chicken gut microbiome. In this study, the spatiotemporal diversity of the gastrointestinal microbiota, shifts in taxonomic composition, and relative abundances of the main bacterial taxa were characterized in Kadaknath, a high-value indigenous Indian chicken breed, using sequencing of the V3–V4 region 16S rRNA gene. To assess microbiome composition and bacterial abundance shifts, three chickens per growth phase (3, 28, and 35 days) were sampled, with microbiota analyzed from three gut regions (crop, small intestine, and ceca) per bird. The results revealed Firmicutes as the most abundant phylum and Lactobacillus as the dominant genus across all stages. Lactobacillus was particularly abundant in the crop at early stages (3 and 28 days), while the ceca exhibited a transition towards the dominance of genus Phocaeicola by day 35. Microbial richness and evenness increased with age, reflecting microbiome maturation, and the analyses of the microbial community composition revealed distinct spatiotemporal differences, with the ceca on day 35 showing the highest differentiation. Pathogen analysis highlighted a peak in poultry-associated taxa Campylobacter, Staphylococcus, and Clostridium paraputrificum in 3-day-old Kadaknath, particularly in the small intestine, underscoring the vulnerability of early growth stages. These findings provide critical insights into age-specific microbiome development and early life-stage susceptibility to pathogens, emphasizing the need for targeted interventions to optimize poultry health management and growth performance.
Pilleriin Peets, Aristeidis Litos, Kai Duehrkop, Daniel Rios Garza, Justin J.J. van der Hooft, Sebastian Boecker, and Bas E. Dutilh (2025) "Chemistry-based vectors map the chemical space of natural biomes from untargeted mass spectrometry", BioRXiv, doi: 10.1101/2025.01.22.634253.
Untargeted metabolomics can comprehensively map the chemical space of a biome, but is limited by low annotation rates (<10%). We used chemistry-based vectors, consisting of molecular fingerprints or chemical compound classes, predicted from mass spectrometry data, to characterize compounds and samples. These chemical characteristics vectors (CCVs) estimate the fraction of compounds with specific chemical properties in a sample. Unlike the aligned MS1 data with intensity information, CCVs incorporate actual chemical properties of compounds, offering deeper insights into sample comparisons. Thus, we identified key compound classes differentiating biomes, such as ethers which are enriched in environmental biomes, while steroids enriched in animal host-related biomes. In biomes with greater variability, CCVs revealed key clustering compound classes, such as organonitrogen compounds in animal distal gut and lipids in animal secretions. CCVs thus enhance the interpretation of untargeted metabolomic data, providing a quantifiable and generalizable understanding of the chemical space of natural biomes.
Álvaro Escobar Doncel, Constantinos Patinios, Alexandre Campos, Maria Beatriz Walter Costa, Maria V. Turkina, Maria Murace, Raymond H.J. Staals, Silvia Vignolini, Bas E. Dutilh, and Colin J. Ingham (2025) "Deletion of the moeA gene in Flavobacterium IR1 drives structural color shift from green to blue and alters polysaccharide metabolism", eLife, doi: 10.7554/eLife.105029.1.
Structural colors (SC), generated by light interacting with nanostructured materials, is responsible for the brightest and most vivid coloration in nature. Despite being widespread within the tree of life, there is little knowledge of the genes involved. Partial exceptions are some Flavobacteriia in which genes involved in a number of pathways, including gliding motility and polysaccharide metabolism, have been linked to SC. A previous genomic analysis of SC and non-SC bacteria suggested that the pterin pathway is involved in the organization of bacteria to form SC. Thus here, we focus on the moeA molybdopterin molybdenum transferase. When this gene was deleted from Flavobacterium IR1, the knock-out mutant showed a strong blue shift in SC of the colony, different from the green SC of the wild-type. The moeA mutant showed a particularly strong blue shift when grown on kappa-carrageenan and was upregulated for starch degradation. To further analyze the molecular changes, proteomic analysis was performed, showing the upregulation of various polysaccharide utilization loci, which supported the link between moeA and polysaccharide metabolism in SC. Overall, we demonstrated that single-gene mutations could change the optical properties of bacterial SC, which is unprecedented when compared to multicellular organisms where structural color is the result of several genes and can not yet be addressed genetically.
Raphaela Joos, Katy Boucher, Aonghus Lavelle, Manimozhiyan Arumugam, Martin J. Blaser, Marcus J. Claesson, Gerard Clarke, Paul D. Cotter, Luisa De Sordi, Maria G. Dominguez-Bello, Bas E. Dutilh, Stanislav D. Ehrlich, Tarini Shankar Ghosh, Colin Hill, Christophe Junot, Leo Lahti, Trevor D. Lawley, Tine R. Licht, Emmanuelle Maguin, Thulani P. Makhalanyane, Julian R. Marchesi, Jelle Matthijnssens, Jeroen Raes, Jacques Ravel, Anne Salonen, Pauline D. Scanlan, Andrey Shkoporov, Catherine Stanton, Ines Thiele, Igor Tolstoy, Jens Walter, Bo Yang, Natalia Yutin, Alexandra Zhernakova, Hub Zwart, Human Microbiome Action Consortium, Joël Doré, and R. Paul Ross (2025) "Examining the healthy human microbiome concept", Nature Reviews Microbiology 23: 192-205, doi: 10.1038/s41579-024-01107-0 Pubmed.
Human microbiomes are essential to health throughout the lifespan and are increasingly recognized and studied for their roles in metabolic, immunological and neurological processes. Although the full complexity of these microbial communities is not fully understood, their clinical and industrial exploitation is well advanced and expanding, needing greater oversight guided by a consensus from the research community. One of the most controversial issues in microbiome research is the definition of a 'healthy' human microbiome. This concept is complicated by the microbial variability over different spatial and temporal scales along with the challenge of applying a unified definition to the spectrum of healthy microbiome configurations. In this Perspective, we examine the progress made and the key gaps that remain to be addressed to fully harness the benefits of the human microbiome. We propose a road map to expand our knowledge of the microbiome-health relationship, incorporating epidemiological approaches informed by the unique ecological characteristics of these communities.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Peter Simmonds, Evelien M. Adriaenssens, Elliot J. Lefkowitz, Hanna M. Oksanen, Stuart G. Siddell, Francisco Murilo Zerbini, Poliane Alfenas-Zerbini, Frank O. Aylward, Donald M. Dempsey, Bas E. Dutilh, Juliana Freitas-Astúa, María Laura García, R. Curtis Hendrickson, Holly R. Hughes, Sandra Junglen, Mart Krupovic, Jens H. Kuhn, Amy J. Lambert, Małgorzata Łobocka, Arcady R. Mushegian, Judit Penzes, Alejandro Reyes Muñoz, David L. Robertson, Simon Roux, Luisa Rubino, Sead Sabanadzovic, Donald B. Smith, Nobuhiro Suzuki, Dann Turner, Koenraad Van Doorslaer, Anne-Mieke Vandamme, and Arvind Varsani (2024) "Changes to virus taxonomy and the ICTV Statutes ratified by the International Committee on Taxonomy of Viruses (2024)"; Archives of Virology 169: 236, doi: 10.1007/s00705-024-06143-y, Pubmed.
This article reports changes to virus taxonomy and taxon nomenclature that were approved and ratified by the International Committee on Taxonomy of Viruses (ICTV) in April 2024. The entire ICTV membership was invited to vote on 203 taxonomic proposals that had been approved by the ICTV Executive Committee (EC) in July 2023 at the 55th EC meeting in Jena, Germany, or in the second EC vote in November 2023. All proposals were ratified by online vote. Taxonomic additions include one new phylum (Ambiviricota), one new class, nine new orders, three new suborders, 51 new families, 18 new subfamilies, 820 new genera, and 3547 new species (excluding taxa that have been abolished). Proposals to complete the process of species name replacement to the binomial (genus + species epithet) format were ratified. Currently, a total of 14,690 virus species have been established.
Meine D. Boer, Chrats Melkonian, Haris Zafeiropoulos, Andreas F. Haas, Daniel Garza, and Bas E. Dutilh (2024) "Improving genome-scale metabolic models of incomplete genomes with deep learning", iScience: 111349, doi: 10.1016/j.isci.2024.111349, Pubmed.
Deciphering microbial metabolism is essential for understanding ecosystem functions. Genome-scale metabolic models (GSMMs) predict metabolic traits from genomic data, but constructing GSMMs for uncultured bacteria is challenging due to incomplete metagenome-assembled genomes, resulting in many gaps. We introduce the Deep Neural Network Guided Imputation Of Reactomes (DNNGIOR), which uses AI to improve gap-filling by learning from the presence and absence of metabolic reactions across diverse bacterial genomes. Key factors for prediction accuracy are: 1) reaction frequency across all bacteria and 2) phylogenetic distance of the query to the training genomes. DNNGIOR predictions achieve an average F1 score of 0.85 for reactions present in over 30% of training genomes. DNNGIOR guided gap-filling was 14 times more accurate for draft reconstructions and 2-9 times for curated models than unweighted gap-filling. DNNGIOR is available at https://github.com/MGXlab/DNNGIOR and as a pip package https://pypi.org/project/dnngior/).
Rick Beeloo, Aldert L. Zomer, Sebastian Deorowicz, and Bas E. Dutilh (2024) "Graphite: painting genomes using a colored de Bruijn graph" NAR Genomics and Bioinformatics 6: lqae142, doi: 10.1093/nargab/lqae142, Pubmed.
The recent growth of microbial sequence data allows comparisons at unprecedented scales, enabling the tracking of strains, mobile genetic elements, or genes. Querying a genome against a large reference database can easily yield thousands of matches that are tedious to interpret and pose computational challenges. We developed Graphite that uses a colored de Bruijn graph (cDBG) to paint query genomes, selecting the local best matches along the full query length. By focusing on the best genomic match of each query region, Graphite reduces the number of matches while providing the most promising leads for sequence tracking or genomic forensics. When applied to hundreds of Campylobacter genomes we found extensive gene sharing, including a previously undetected C. coli plasmid that matched a C. jejuni chromosome. Together, genome painting using cDBGs as enabled by Graphite, can reveal new biological phenomena by mitigating computational hurdles.
Yasas Wijesekara, Ling-Yi Wu, Rick Beeloo, Piotr Rozwalak, Ernestina Hauptfeld, Swapnil Prakash Doijad, Bas E. Dutilh, and Lars Kaderali (2024) "Jaeger: an accurate and fast deep-learning tool to detect bacteriophage sequences", BioRXiv, doi: 10.1101/2024.09.24.612722.
Viruses are integral to every biome on Earth, yet we still need a more comprehensive picture of their identity and global distribution. Global metagenomics sequencing efforts revealed the genomic content of tens of thousands of environmental samples, however identifying the viral sequences in these datasets remains challenging due to their vast genomic diversity. Here, we address identifying bacteriophage sequences in sequencing data. In a recent benchmarking paper, we observed that existing deep-learning tools show a high true positive rate, but often produce many false positives when confronted with divergent sequences. To tackle this challenge, we introduce Jaeger, a novel deep-learning method designed specifically for identifying bacteriophage genome fragments. Extensive benchmarking on the IMG/VR database and real-world metagenomes reveals Jaeger's consistent performance across various scenarios. Applying Jaeger to over 16,000 metagenomic assemblies from the MGnify database yielded over five million putative phage contigs at an estimated 2-27% false discovery rate. On average, Jaeger is around 20 times faster than the other state-of-the-art methods, highlighting its efficacy in bacteriophage identification within global metagenomes. Jaeger is available at https://github.com/MGXlab/Jaeger.
Piotr Rozwalak, Bas E. Dutilh, Andrzej Zielezinski, and others (2024) "How much do viruses change over time?" Biomedical Science Journal for Kids.
Bacteriophages, or just phages, are tiny viruses that infect bacteria. Some phages help us by killing harmful bacteria and keeping our gut healthy. Although phages have existed for millions of years, we don't know much about the ones from ancient times. To find out, we analyzed super old human poop samples. Using a new technique called de novo assembly, we pieced together the DNA of ancient viruses from these samples. We discovered nearly 300 types of bacteriophages that we had never seen before. But there was also one 1,300-year-old phage that was almost exactly the same as a modern day species. This was extremely surprising! Our research provides a useful method to rebuild ancient phage DNA sequences and learn more about the history of viruses.
Patrick A. de Jonge, Bert-Jan H. van den Born, Aeilko H. Zwinderman, Max Nieuwdorp Bas E. Dutilh, and Hilde Herrema (2024) "Phylogeny and disease associations of a widespread and ancient intestinal bacteriophage lineage", Nature Communications 15: 6346, doi: 10.1038/s41467-024-50777-0, Pubmed.
Viruses are core components of the human microbiome, impacting health through interactions with gut bacteria and the immune system. Most human microbiome viruses are bacteriophages, which exclusively infect bacteria. Until recently, most gut virome studies focused on low taxonomic resolution (e.g., viral operational taxonomic units), hampering population-level analyses. We previously identified an expansive and widespread bacteriophage lineage in inhabitants of Amsterdam, the Netherlands. Here, we study their biodiversity and evolution in various human populations. Based on a phylogeny using sequences from six viral genome databases, we propose the Candidatus order Heliusvirales. We identify heliusviruses in 82% of 5441 individuals across 39 studies, and in nine metagenomes from humans that lived in Europe and North America between 1000 and 5000 years ago. We show that a large lineage started to diversify when Homo sapiens first appeared some 300,000 years ago. Ancient peoples and modern hunter-gatherers have distinct Ca. Heliusvirales populations with lower richness than modern urbanized people. Urbanized people suffering from type 1 and type 2 diabetes, as well as inflammatory bowel disease, have higher Ca. Heliusvirales richness than healthy controls. We thus conclude that these ancient core members of the human gut virome have thrived with increasingly westernized lifestyles.
Abraham L. van Eijnatten, Luc van Zon, Eleni Manousou, Margarita Bikineeva, Jasper Wubs, Wim van der Putten, Elly Morriën, Bas E. Dutilh, and Basten L. Snoek (2024) "SpeSpeNet: An interactive and user-friendly tool to create and explore microbial correlation networks", BioRXiv, doi: 10.1101/2024.07.17.603889.
Correlation networks are commonly used to explore microbiome data. In these networks, nodes are taxa and edges represent correlations between their abundance patterns across samples. As clusters of correlating taxa (co-abundance clusters) often indicate a shared response to environmental drivers, network visualization contributes to system understanding. Currently, most tools for creating and visualizing co-abundance networks from microbiome data either require the researcher to have coding skills, or they are not user-friendly, with high time expenditure and limited customizability. Furthermore, existing tools lack focus on the relationship between environmental drivers and the structure of the microbiome, even though many edges in correlation networks can be understood through a shared relationship of two taxa with the environment. For these reasons we developed SpeSpeNet (Species-Species Network, https://tbb.bio.uu.nl/SpeSpeNet), a practical and user-friendly R-shiny tool to construct and visualize correlation networks from taxonomic abundance tables. The details of data preprocessing, network construction, and visualization are automated, require no programming ability for the web version, and are highly customizable, including associations with user-provided environmental data. Here, we present the details of SpeSpeNet and demonstrate its utility using three case studies.
Aldert Zomer, Colin J. Ingham, F.A. Bastiaan von Meijenfeldt, Álvaro Escobar Doncel, Gea T. van de Kerkhof, Raditijo Hamidjaja, Sanne Schouten, Lukas Schertel, Karin H. Müller, Laura Catón, Richard L. Hahnke, Henk Bolhuis, Silvia Vignolini, and Bas E. Dutilh (2024) "Structural color in the bacterial domain: the ecogenomics of a 2-dimensional optical phenotype", PNAS 121: e2309757121, doi: 10.1073/pnas.230975712, Pubmed. Access Science, FSU, Microverse, MPI, NIOZ, Phys.org, Populat Science, UU.
Structural color is an optical phenomenon resulting from light interacting with nanostructured materials. Although structural color (SC) is widespread in the tree of life, the underlying genetics and genomics are not well understood. Here, we collected and sequenced a set of 87 structurally colored bacterial isolates and 30 related strains lacking SC. Optical analysis of colonies indicated that diverse bacteria from at least two different phyla (Bacteroidetes and Proteobacteria) can create two-dimensional packing of cells capable of producing SC. A pan-genome-wide association approach was used to identify genes associated with SC. The biosynthesis of uroporphyrin and pterins, as well as carbohydrate utilization and metabolism, was found to be involved. Using this information, we constructed a classifier to predict SC directly from bacterial genome sequences and validated it by cultivating and scoring 100 strains that were not part of the training set. We predicted that SC is widely distributed within gram-negative bacteria. Analysis of over 13,000 assembled metagenomes suggested that SC is nearly absent from most habitats associated with multicellular organisms except macroalgae and is abundant in marine waters and surface/air interfaces. This work provides a large-scale ecogenomics view of SC in bacteria and identifies microbial pathways and evolutionary relationships that underlie this optical phenomenon.
Sanne W.M. Poppeliers, Juan J. Sánchez-Gil, José L. López, Bas E. Dutilh, Corné M.J. Pieterse, and Ronnie de Jonge (2024) "High-resolution quantification of the rhizosphere effect along a soil-to-root gradient shows selection-driven convergence of rhizosphere microbiomes", BioRXiv, doi: 10.1101/2024.06.21.600027v1.
Plants secrete a complex array of organic compounds, constituting about a third of their photosynthetic products, into the surrounding soil. As a result, concentration gradients are established from the roots into the bulk soil, known as the rhizosphere. Soil microbes benefit from these root exudates for their survival and propagation, and consequently, the composition of the rhizosphere microbial community follows the gradient of available compounds, a phenomenon oftentimes referred to as the rhizosphere effect. However, the fine-grained changes in the microbial community along this soil-root gradient have not been well described. Yet such insights would enable us to underpin the ecological rules underlying root microbial community assembly. Therefore, here we harvested the roots of individual Arabidopsis thaliana plants grown in three different natural soils at high-resolution, such that we could interrogate community assembly and predict microbial growth rate across consecutive, fine-grained, rhizosphere compartments. We found that the strength of the rhizosphere effect depends on root proximity and that microbial communities closer to the roots harbour related microbes. Closer to the roots, microbial community assembly became less random and more driven by selection-based processes. Intriguingly, we observed priority effects, where related microbes that arrive first are more likely to establish, and that microbes might use different ecological growth strategies to colonise the rhizosphere. All effects appeared to be independent from starting conditions as microbial community composition converged on the root despite different soil microbial seed banks. Together, our results provide a high-resolution view of the microbiome changes across the soil-root gradient.
Arista Fourie, José L. López, Juan J. Sánchez-Gil, Sanne W.M. Poppeliers, Ronnie de Jonge, and Bas E. Dutilh (2024) "Bacterial family-specific enrichment and functions of secretion systems in the rhizosphere", BioRXiv, doi: 10.1101/2024.05.07.592589.
The plant rhizosphere is a highly selective environment where bacteria have developed traits to establish themselves or outcompete other microbes. These traits include bacterial secretion systems (SSs) that range from Type I (T1SS) to Type IX (T9SS) and can play diverse roles. The best known functions are to secrete various proteins or other compounds into the extracellular space or into neighbouring cells, including toxins to attack other microbes or effectors to suppress plant host immune responses. Here, we aimed to determine which bacterial SS's were associated with the plant rhizosphere. We utilised paired metagenomic datasets of rhizosphere and bulk soil samples from five different plant species grown in a wide variety of soil types, amounting to ten different studies. The T3SS and T6SS were generally enriched in the rhizosphere, as observed in studies of individual plant-associated genera. We also identified additional SS's that have received less attention thus far, such as the T2SS, T5SS and Bacteroidetes-specific T6SSiii and T9SS. The predicted secreted proteins of some of these systems (T3SS, T5SS and T6SS) could be linked to functions such as toxin secretion, adhesion to the host and facilitation of plant-host interactions (such as root penetration). The most prominent bacterial taxa with rhizosphere- or soil-enriched SS's included Xanthomonadaceae, Oxalobacteraceae, Comamonadaceae, Caulobacteraceae, and Chitinophagaceae, broadening the scope of known plant-associated taxa that use these systems. We anticipate that the SSs and taxa identified in this study may be utilised for the optimisation of bioinoculants to improve plant productivity.
Gonçalo Piedade, Max E. Schön, Cédric Lood, Mikhail V. Fofanov, Ella M. Wesdorp, Tristan E.G. Biggs, Ling-Yi Wu, Henk Bolhuis, Matthias G. Fischer, Natalya Yutin, Bas E. Dutilh, and Corina Brussaard (2024) "Seasonal dynamics and diversity of Antarctic marine viruses reveal a novel viral seascape", Nature Communications 15: 9192, doi: 10.1038/s41467-024-53317-y, Pubmed. NIOZ, Behind the Paper.
The Southern Ocean microbial ecosystem, with its pronounced seasonal shifts, is vulnerable to the impacts of climate change. Since viruses are key modulators of microbial abundance, diversity, and evolution, we need a better understanding of the effects of seasonality on the viruses in this region. Our comprehensive exploration of DNA viral diversity in the Southern Ocean reveals a unique and largely uncharted viral landscape, of which 75% was previously unidentified in other oceanic areas. We uncover novel viral taxa at high taxonomic ranks, expanding our understanding of crassphage, polinton-like virus, and virophage diversity. Nucleocytoviricota viruses represent an abundant and diverse group of Antarctic viruses, highlighting their potential as important regulators of phytoplankton population dynamics. Our temporal analysis reveals complex seasonal patterns in marine viral communities (bacteriophages, eukaryotic viruses) which underscores the apparent interactions with their microbial hosts, whilst deepening our understanding of their roles in the world's most sensitive and rapidly changing ecosystem.
Ernestina Hauptfeld, Nikolaos Pappas, Sandra van Iwaarden, Basten L. Snoek, Andrea Aldas-Vargas, Bas E. Dutilh, and F.A. Bastiaan von Meijenfeldt (2024) "Integrating taxonomic signals from MAGs and contigs improves read annotation and taxonomic profiling of metagenomes", Nature Communications 15: 3373, doi: 10.1038/s41467-024-47155-1, Pubmed.
Metagenomic analysis typically includes read-based taxonomic profiling, assembly, and binning of metagenome-assembled genomes (MAGs). Here we integrate these steps in Read Annotation Tool (RAT), which uses robust taxonomic signals from MAGs and contigs to enhance read annotation. RAT reconstructs taxonomic profiles with high precision and sensitivity, outperforming other state-of-the-art tools. In high-diversity groundwater samples, RAT annotates a large fraction of the metagenomic reads, calling novel taxa at the appropriate, sometimes high taxonomic ranks. Thus, RAT integrative profiling provides an accurate and comprehensive view of the microbiome from shotgun metagenomics data. The package of Contig Annotation Tool (CAT), Bin Annotation Tool (BAT), and RAT is available at https://github.com/MGXlab/CAT_pack (from CAT pack v6.0). The CAT pack now also supports Genome Taxonomy Database (GTDB) annotations.
Ling-Yi Wu, Yasas Wijesekara, Gonçalo Piedade, Nikolaos Pappas, Corina P.D. Brussaard, and Bas E. Dutilh (2024) "Benchmarking bioinformatic virus identification tools using real-world metagenomic data across biomes", Genome Biology 25: 97, doi: 10.1186/s13059-024-03236-4, Pubmed.
Background As most viruses remain uncultivated, metagenomics is currently the main method for virus discovery. Detecting viruses in metagenomic data is not trivial. In the past few years, many bioinformatic virus identification tools have been developed for this task, making it challenging to choose the right tools, parameters, and cutoffs. As all these tools measure different biological signals, and use different algorithms and training and reference databases, it is imperative to conduct an independent benchmarking to give users objective guidance. Results We compare the performance of nine state-of-the-art virus identification tools in thirteen modes on eight paired viral and microbial datasets from three distinct biomes, including a new complex dataset from Antarctic coastal waters. The tools have highly variable true positive rates (0-97%) and false positive rates (0-30%). PPR-Meta best distinguishes viral from microbial contigs, followed by DeepVirFinder, VirSorter2, and VIBRANT. Different tools identify different subsets of the benchmarking data and all tools, except for Sourmash, find unique viral contigs. Performance of tools improved with adjusted parameter cutoffs, indicating that adjustment of parameter cutoffs before usage should be considered. Conclusions Together, our independent benchmarking facilitates selecting choices of bioinformatic virus identification tools and gives suggestions for parameter adjustments to viromics researchers.
Daniel Loos, Ailton Pereira da Costa Filho, Bas E. Dutilh, Amelia E. Barber, and Gianni Panagiotou (2024) "A global survey of host, aquatic, and soil microbiomes reveals shared abundance and genomic features between bacterial and fungal generalists", Cell Reports 43: 114046, doi: 10.1016/j.celrep.2024.114046, Pubmed.
Environmental change, coupled with alteration in human lifestyles, is profoundly impacting the microbial communities critical to the health of the Earth and its inhabitants. To identify bacteria and fungi that are resistant and susceptible to habitat change, we analyze thousands of genera detected in 1,580 host, soil, and aquatic samples. This large-scale analysis identifies 48 bacterial and 4 fungal genera that are abundant across the three biomes, demonstrating fitness in diverse environmental conditions. Samples containing these generalists have significantly higher alpha diversity. These generalists play a significant role in shaping cross-kingdom community structure, boasting larger genomes with more secondary metabolism and antimicrobial resistance genes. Conversely, 30 bacterial and 19 fungal genera are only found in a single habitat, suggesting a limited ability to adapt to different and changing environments. These findings contribute to our understanding of microbial niche breadth and its consequences for global biodiversity loss.
Katarina Hrovat, Bas E. Dutilh, Marnix H. Medema, and Chrats Melkonian (2024) "Taxonomic resolution of different 16S rRNA variable regions varies strongly across plant-associated bacteria", ISME Communications 4: ycae034, doi: 10.1093/ismeco/ycae034, Pubmed.
Plant-microbiome research plays a pivotal role in understanding the relationships between plants and their associated microbial communities, with implications for agriculture and ecosystem dynamics. Metabarcoding analysis on variable regions of the 16S ribosomal RNA (rRNA) gene remains the dominant technology to study microbiome diversity in this field. However, the choice of the targeted variable region might affect the outcome of the microbiome studies. In our in-silico analysis, we have evaluated whether the targeted variable region has an impact on taxonomic resolution in 16 plant-related microbial genera. Through a comparison of 16S rRNA gene variable regions with whole-genome data, our findings suggest that the V1-V3 region is generally a more suitable option than the widely used V3-V4 region for targeting microbiome analysis in plant-related genera. However, sole reliance on one region could introduce detection biases for specific genera. Thus, we are suggesting that while transitioning to full-length 16S rRNA gene and whole-genome sequencing for plant-microbiome analysis, the usage of genus-specific variable regions can achieve more precise taxonomic assignments. More broadly, our approach provides a blueprint to identify the most discriminating variable regions of the 16S rRNA gene for genus of interest.
Piotr Rozwalak, Jakub Barylski, Yasas Wijesekara, Bas E. Dutilh, and Andrzej Zielezinski (2024) "Ultraconserved bacteriophage genome sequence identified in 1300-year-old human palaeofaeces", Nature Communications 15: 495, doi: 10.1038/s41467-023-44370-0, Pubmed.
Bacteriophages are widely recognised as rapidly evolving biological entities. However, knowledge about ancient bacteriophages is limited. Here, we analyse DNA sequence datasets previously generated from ancient palaeofaeces and human gut-content samples, and identify an ancient phage genome nearly identical to present-day Mushuvirus mushu, a virus that infects gut commensal bacteria. The DNA damage patterns of the genome are consistent with its ancient origin and, despite 1300 years of evolution, the ancient Mushuvirus genome shares 97.7% nucleotide identity with its modern counterpart, indicating a long-term relationship between the prophage and its host. In addition, we reconstruct and authenticate 297 other phage genomes from the last 5300 years, including those belonging to unknown families. Our findings demonstrate the feasibility of reconstructing ancient phage genome sequences, thus expanding the known virosphere and offering insights into phage-bacteria interactions spanning several millennia.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Jeroen Meijer, Petros Skiadas, Paul B. Rainey, Paulien Hogeweg, and Bas E. Dutilh (2023) "Eco-evolutionary dynamics of massive, parallel bacteriophage outbreaks in compost communities", BioRXiv, doi: 10.1101/2023.07.31.550844.
Bacteriophages are important drivers of microbial ecosystems, but their influence and dynamics in terrestrial biomes remain poorly understood compared to aquatic and host-associated systems. To investigate this, we analyzed shotgun metagenomics datasets from ten compost-derived microbial communities propagated over 48 weeks. We found that the communities clustered into two distinct types consisting of hundreds of microbial genera, and in one community type identified Theomophage, a lytic bacteriophage representing a new Schitoviridae subfamily, which accounted for up to 74.3% of the total community metagenome, indicating massive viral outbreaks. We tracked molecular evolution of Theomophage and found that isolated communities were dominated by a single strain that showed little molecular evolution during outbreaks. However, when experimental manipulation allowed phages to migrate between communities, we observed transient coexistence of strains followed by genomic recombination that underpinned replacement of the ancestral strains. Additionally, when Theomophage colonized mesocosms where it was originally absent, new mutations evolved that fixed and spread to other communities. Our study describes the largest bacteriophage outbreak reported to date and reveals the spatial and temporal scales at which terrestrial bacteriophage microdiversity evolves. It also demonstrates that mixing of viral communities, which may be frequent in natural systems, promotes rapid bacteriophage evolution.
Bram van Dijk, Pauline Buffard, Andrew D. Farr, Franz Giersdorf, Jeroen Meijer, Bas E. Dutilh, and Paul B. Rainey (2023) "Identifying and tracking mobile elements in evolving compost communities yields insights into the nanobiome", ISME Communications 3: 90, doi: 10.1038/s43705-023-00294-w. Pubmed.
Evelien M. Adriaenssens, Simon Roux, J. Rodney Brister, Ilene Karsch-Mizrachi, Jens H. Kuhn, Arvind Varsani, Tong Yigang, Alejandro Reyes, Cédric Lood, Elliot J. Lefkowitz, Matthew B. Sullivan, Robert A. Edwards, Peter Simmonds, Luisa Rubino, Sead Sabanadzovic, Mart Krupovic, and Bas E. Dutilh (2023) "Guidelines for public database submission of uncultivated virus genome sequences for taxonomic classification", Nature Biotechnology 41: 898-902, doi: 10.1038/s41587-023-01844-2. Pubmed.
José L. López, Arista Fourie, Sanne W. M. Poppeliers, Nikolaos Pappas, Juan J. Sánchez-Gil, Ronnie de Jonge, and Bas E. Dutilh (2023) "Growth rate is a dominant factor predicting the rhizosphere effect", ISME Journal 17: 1396-1405, doi: 10.1038/s41396-023-01453-6. Pubmed. Behind the paper.
Francisco Murilo Zerbini, Stuart G. Siddell, Elliot J. Lefkowitz, Arcady R. Mushegian, Evelien M. Adriaenssens, Poliane Alfenas-Zerbini, Donald M. Dempsey, Bas E. Dutilh, María Laura García, R. Curtis Hendrickson, Sandra Junglen, Mart Krupovic, Jens H. Kuhn, Amy J. Lambert, Małgorzata Łobocka, Hanna M. Oksanen, David L. Robertson, Luisa Rubino, Sead Sabanadzovic, Peter Simmonds, Donald B. Smith, Nobuhiro Suzuki, Koenraad Van Doorslaer, Anne-Mieke Vandamme, and Arvind Varsani (2023) "Changes to virus taxonomy and the ICTV Statutes ratified by the International Committee on Taxonomy of Viruses (2023)", Archives of Virology 168: 175, doi: 10.1007/s00705-023-05797-4. Pubmed.
Carlijn E. Bruggeling, Maarten Te Groen, Daniel R. Garza, Famke van Heeckeren Tot Overlaer, Joyce P.M. Krekels, Basma-Chick Sulaiman, Davy Karel, Athreyu Rulof, Anne R. Schaaphok, Daniel L.A.H. Hornikx, Iris D. Nagtegaal, Bas E. Dutilh, Frank Hoentjen, and Annemarie Boleij (2023) "Bacterial oncotraits rather than spatial organization are associated with dysplasia in ulcerative colitis", Journal of Crohn's and colitis 17: 1870-1881, doi: 10.1093/ecco-jcc/jjad092. Pubmed.
| Background and aims: Colonic bacterial biofilms are frequently present in ulcerative colitis (UC) and may increase dysplasia risk through pathogens expressing oncotraits. This prospective cohort study aimed to determine (1) the association of oncotraits and longitudinal biofilm presence with dysplasia risk in UC, and (2) the relation of bacterial composition with biofilms and dysplasia risk. Methods: Feces and left- and right-sided colonic biopsies were collected from 80 UC patients and 35 controls. Oncotraits (FadA of Fusobacterium, BFT of Bacteroides fragilis, colibactin (ClbB) and Intimin (Eae) of Escherichia coli) were assessed in fecal DNA with multiplex qPCR. Biopsies were screened for biofilms (n=873) with 16S rRNA fluorescent in situ hybridization. Shotgun metagenomic sequencing (n=265), and ki67-immunohistochemistry were performed. Associations were determined with a mixed-effects regression model. Results: Biofilms were highly prevalent in UC patients (90.8%) with a median persistence of 3 years (IQR 2-5 years). Biofilm-positive biopsies showed increased epithelial hypertrophy (p=0.025), a reduced Shannon diversity independent of disease status (p=0.015), however, were not significantly associated with dysplasia in UC (aOR 1.45(95%CI0.63-3.40). In contrast, ClbB independently associated with dysplasia (aOR 7.16 (95%CI1.75-29.28), while FadA and Fusobacteriales were associated with a decreased dysplasia risk in UC (aOR 0.23 (95%CI0.06-0.83), and p<0.01). Conclusions: Biofilms are a hallmark of UC, however, because of their high prevalence a poor biomarker for dysplasia. In contrast, colibactin presence and FadA absence independently associate with dysplasia in UC and might therefore be valuable biomarkers for future risk stratification and intervention strategies. |

|
Simon Roux, Antonio Pedro Camargo, Felipe H. Coutinho, Shareef M. Dabdoub, Bas E. Dutilh, Stephen Nayfach, and Andrew Tritt (2023), "iPHoP: An integrated machine learning framework to maximize host prediction for metagenome-derived viruses of archaea and bacteria", PLoS Biology 21: e3002083, doi: 10.1371/journal.pbio.3002083, Pubmed.
The extraordinary diversity of viruses infecting bacteria and archaea is now primarily studied through metagenomics. While metagenomes enable high-throughput exploration of the viral sequence space, metagenome-derived sequences lack key information compared to isolated viruses, in particular host association. Different computational approaches are available to predict the host(s) of uncultivated viruses based on their genome sequences, but thus far individual approaches are limited either in precision or in recall, i.e., for a number of viruses they yield erroneous predictions or no prediction at all. Here, we describe iPHoP, a two-step framework that integrates multiple methods to reliably predict host taxonomy at the genus rank for a broad range of viruses infecting bacteria and archaea, while retaining a low false discovery rate. Based on a large dataset of metagenome-derived virus genomes from the IMG/VR database, we illustrate how iPHoP can provide extensive host prediction and guide further characterization of uncultivated viruses.
Stuart G. Siddell, Donald B. Smith, Evelien Adriaenssens, Poliane Alfenas-Zerbini, Bas E. Dutilh, Maria Laura Garcia, Sandra Junglen, Mart Krupovic, Jens H. Kuhn, Amy J. Lambert, Elliot J. Lefkowitz, Małgorzata Łobocka, Arcady R. Mushegian, Hanna M. Oksanen, David L. Robertson, Luisa Rubino, Sead Sabanadzovic, Peter Simmonds, Nobuhiro Suzuki, Koenraad Van Doorslaer, Anne-Mieke Vandamme, Arvind Varsani, and F. Murilo Zerbini (2023), "Virus taxonomy and the role of the International Committee on Taxonomy of Viruses (ICTV)", Journal of General Virology 104: 001840, doi: 10.1099/jgv.0.001840, Pubmed.
The taxonomy of viruses is developed and overseen by the International Committee on Taxonomy of Viruses (ICTV), which scrutinizes, approves and ratifies taxonomic proposals, and maintains a list of virus taxa with approved names (https://ictv.global). The ICTV has approximately 180 members who vote by simple majority. Taxon-specific Study Groups established by the ICTV have a combined membership of over 600 scientists from the wider virology community; they provide comprehensive expertise across the range of known viruses and are major contributors to the creation and evaluation of taxonomic proposals. Proposals can be submitted by anyone and will be considered by the ICTV irrespective of Study Group support. Thus, virus taxonomy is developed from within the virology community and realized by a democratic decision-making process. The ICTV upholds the distinction between a virus or replicating genetic element as a physical entity and the taxon category to which it is assigned. This is reflected by the nomenclature of the virus species taxon, which is now mandated by the ICTV to be in a binomial format (genus + species epithet) and is typographically distinct from the names of viruses. Classification of viruses below the rank of species (such as, genotypes or strains) is not within the remit of the ICTV. This article, authored by the ICTV Executive Committee, explains the principles of virus taxonomy and the organization, function, processes and resources of the ICTV, with the aim of encouraging greater understanding and interaction among the wider virology community.
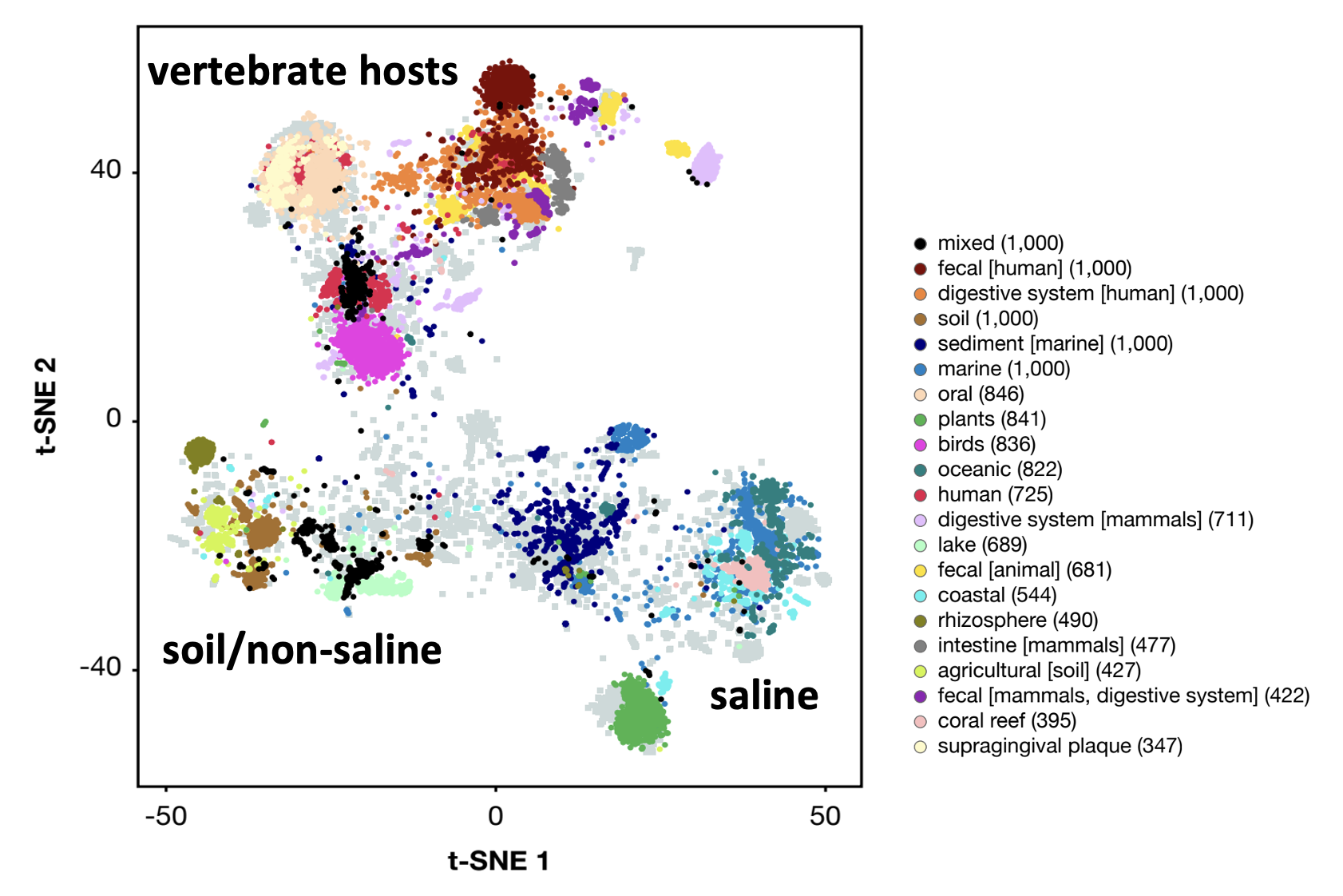
F.A. Bastiaan von Meijenfeldt, Paulien Hogeweg, and Bas E. Dutilh (2023), "A social niche breadth score reveals niche range strategies of generalists and specialists", Nature Ecology and Evolution 7: 768-781, doi: 10.1038/s41559-023-02027-7, Pubmed. Press: Nature E&E News & Views, UU, FSU.
|
Generalists can survive in many environments, whereas specialists are restricted to a single environment. Although a classical concept in ecology, niche breadth has remained challenging to quantify for microorganisms because it depends on an objective definition of the environment. Here, by defining the environment of a microorganism as the community it resides in, we integrated information from over 22,000 environmental sequencing samples to derive a quantitative measure of the niche, which we call social niche breadth. At the level of genera, we explored niche range strategies throughout the prokaryotic tree of life. We found that social generalists include opportunists that stochastically dominate local communities, whereas social specialists are stable but low in abundance. Social generalists have a more diverse and open pan-genome than social specialists, but we found no global correlation between social niche breadth and genome size. Instead, we observed two distinct evolutionary strategies, whereby specialists have relatively small genomes in habitats with low local diversity, but relatively large genomes in habitats with high local diversity. Together, our analysis shines data-driven light on microbial niche range strategies. |
|
Peter Simmonds, Evelien M. Adriaenssens, F. Murilo Zerbini, Nicola G.A. Abrescia, Pakorn Aiewsakun, Poliane Alfenas-Zerbini, Yiming Bao, Jakub Barylski, Christian Drosten, Siobain Duffy, W. Paul Duprex, Bas E. Dutilh, Santiago F. Elena, María Laura García, Sandra Junglen, Aris Katzourakis, Eugene V. Koonin, Mart Krupovic, Jens H. Kuhn, Amy J. Lambert, Elliot J. Lefkowitz, Małgorzata Łobocka, Cédric Lood, Jennifer Mahony, Jan P. Meier-Kolthoff, Arcady R. Mushegian, Hanna M. Oksanen, Minna M. Poranen, Alejandro Reyes-Muñoz, David L. Robertson, Simon Roux, Luisa Rubino, Sead Sabanadzovic, Stuart Siddell, Tim Skern, Donald B. Smith, Matthew B. Sullivan, Nobuhiro Suzuki, Dann Turner, Koenraad Van Doorslaer, Anne-Mieke Vandamme, Arvind Varsani, and Nikos Vasilakis (2023), "Four principles to establish a universal virus taxonomy", PLoS Biology 21: e3001922, doi: 10.1371/journal.pbio.3001922, Pubmed.
|
A universal taxonomy of viruses is essential for a comprehensive view of the virus world and for communicating the complicated evolutionary relationships among viruses. However, there are major differences in the conceptualisation and approaches to virus classification and nomenclature among virologists, clinicians, agronomists, and other interested parties. Here, we provide recommendations to guide the construction of a coherent and comprehensive virus taxonomy, based on expert scientific consensus. Firstly, assignments of viruses should be congruent with the best attainable reconstruction of their evolutionary histories, i.e., taxa should be monophyletic. This fundamental principle for classification of viruses is currently included in the International Committee on Taxonomy of Viruses (ICTV) code only for the rank of species. Secondly, phenotypic and ecological properties of viruses may inform, but not override, evolutionary relatedness in the placement of ranks. Thirdly, alternative classifications that consider phenotypic attributes, such as being vector-borne (e.g., "arboviruses"), infecting a certain type of host (e.g., "mycoviruses," "bacteriophages") or displaying specific pathogenicity (e.g., "human immunodeficiency viruses"), may serve important clinical and regulatory purposes but often create polyphyletic categories that do not reflect evolutionary relationships. Nevertheless, such classifications ought to be maintained if they serve the needs of specific communities or play a practical clinical or regulatory role. However, they should not be considered or called taxonomies. Finally, while an evolution-based framework enables viruses discovered by metagenomics to be incorporated into the ICTV taxonomy, there are essential requirements for quality control of the sequence data used for these assignments. Combined, these four principles will enable future development and expansion of virus taxonomy as the true evolutionary diversity of viruses becomes apparent. |
|
Liliane S. Oliveira, Alejandro Reyes, Bas E. Dutilh, and Arthur Gruber (2023), "Rational design of profile HMMs for sensitive and specific sequence detection with case studies applied to viruses, bacteriophages, and casposons", Viruses 15: 519, doi: 1999-4915/15/2/519, Pubmed.
Profile hidden Markov models (HMMs) are a powerful way of modeling biological sequence diversity and constitute a very sensitive approach to detecting divergent sequences. Here, we report the development of protocols for the rational design of profile HMMs. These methods were implemented on TABAJARA, a program that can be used to either detect all biological sequences of a group or discriminate specific groups of sequences. By calculating position-specific information scores along a multiple sequence alignment, TABAJARA automatically identifies the most informative sequence motifs and uses them to construct profile HMMs. As a proof-of-principle, we applied TABAJARA to generate profile HMMs for the detection and classification of two viral groups presenting different evolutionary rates: bacteriophages of the Microviridae family and viruses of the Flavivirus genus. We obtained conserved models for the generic detection of any Microviridae or Flavivirus sequence, and profile HMMs that can specifically discriminate Microviridae subfamilies or Flavivirus species. In another application, we constructed Cas1 endonuclease-derived profile HMMs that can discriminate CRISPRs and casposons, two evolutionarily related transposable elements. We believe that the protocols described here, and implemented on TABAJARA, constitute a generic toolbox for generating profile HMMs for the highly sensitive and specific detection of sequence classes.
Dann Turner, Andrey N. Shkoporov, Cédric Lood, Andrew D. Millard, Bas E. Dutilh, Poliane Alfenas-Zerbini, Leonardo J. van Zyl, Ramy K. Aziz, Hanna M. Oksanen, Minna M. Poranen, Andrew M. Kropinski, Jakub Barylski, J. Rodney Brister, Nina Chanisvili, Rob A. Edwards, François Enault, Annika Gillis, Petar Knezevic, Mart Krupovic, Ipek Kurtböke, Alla Kushkina, Rob Lavigne, Susan Lehman, Malgorzata Lobocka, Cristina Moraru, Andrea Moreno Switt, Vera Morozova, Jesca Nakavuma, Alejandro Reyes Muñoz, Jānis Rūmnieks, B.L. Sarkar, Matthew B. Sullivan, Jumpei Uchiyama, Johannes Wittmann, Tong Yigang, and Evelien M. Adriaenssens (2023), "Abolishment of morphology-based taxa and change to binomial species names: 2022 taxonomy update of the ICTV bacterial viruses subcommittee", Archives of Virology 168: 74, doi: 10.1007/s00705-022-05694-2, Pubmed.
This article summarises the activities of the Bacterial Viruses Subcommittee of the International Committee on Taxonomy of Viruses for the period of March 2021-March 2022. We provide an overview of the new taxa proposed in 2021, approved by the Executive Committee, and ratified by vote in 2022. Significant changes to the taxonomy of bacterial viruses were introduced: the paraphyletic morphological families Podoviridae, Siphoviridae, and Myoviridae as well as the order Caudovirales were abolished, and a binomial system of nomenclature for species was established. In addition, one order, 22 families, 30 subfamilies, 321 genera, and 862 species were newly created, promoted, or moved.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Thomas S. Postler, Luisa Rubino, Evelien M. Adriaenssens, Bas E. Dutilh, Balázs Harrach, Sandra Junglen, Andrew M. Kropinski, Mart Krupovic, Jiro Wada, Anya Crane, Jens H. Kuhn, Arcady Mushegian, Jānis Rūmnieks, Sead Sabanadzovic, Peter Simmonds, Arvind Varsani, F. Murilo Zerbini, Julie Callanan, Lorraine A. Draper, Colin Hill, and Stephen R. Stockdale (2022), "Guidance for creating individual and batch latinized binomial virus species names", Journal of General Virology 103: 001800, doi: 10.1099/jgv.0.001800. Pubmed.
The International Committee on Taxonomy of Viruses recently adopted, and is gradually implementing, a binomial naming format for virus species. Although full Latinization of these names remains optional, a standardized nomenclature based on Latinized binomials has the advantage of comparability with all other biological taxonomies. As a language without living native speakers, Latin is more culturally neutral than many contemporary languages, and words built from Latin roots are already widely used in the language of science across the world. Conversion of established species names to Latinized binomials or creation of Latinized binomials de novo may seem daunting, but the rules for name creation are straightforward and can be implemented in a formulaic manner. Here, we describe approaches, strategies and steps for creating Latinized binomials for virus species without prior knowledge of Latin. We also discuss a novel approach to the automated generation of large batches of novel genus and species names. Importantly, conversion to a binomial format does not affect virus names, many of which are created from local languages.
Peter J. Walker, Stuart G. Siddell, Elliot J. Lefkowitz, Arcady R. Mushegian, Evelien M. Adriaenssens, Poliane Alfenas-Zerbini, Donald M. Dempsey, Bas E. Dutilh, María Laura García, R. Curtis Hendrickson, Sandra Junglen, Mart Krupovic, Jens H. Kuhn, Amy J. Lambert, Małgorzata Łobocka, Hanna M. Oksanen, Richard J. Orton, David L. Robertson, Luisa Rubino, Sead Sabanadzovic, Peter Simmonds, Donald B. Smith, Nobuhiro Suzuki, Koenraad Van Doorslaer, Anne-Mieke Vandamme, Arvind Varsani, and Francisco Murilo Zerbini (2022), "Recent changes to virus taxonomy ratified by the International Committee on Taxonomy of Viruses (2022)", Archives of Virology 167: 2429-2440, doi: 10.1007/s00705-022-05516-5, Pubmed.
This article reports the changes to virus taxonomy approved and ratified by the International Committee on Taxonomy of Viruses (ICTV) in March 2022. The entire ICTV was invited to vote on 174 taxonomic proposals approved by the ICTV Executive Committee at its annual meeting in July 2021. All proposals were ratified by an absolute majority of the ICTV members. Of note, the Study Groups have started to implement the new rule for uniform virus species naming that became effective in 2021 and mandates the binomial 'Genus_name species_epithet' format with or without Latinization. As a result of this ratification, the names of 6,481 virus species (more than 60 percent of all species names currently recognized by ICTV) now follow this format.
Daniel R. Garza, F. A. Bastiaan von Meijenfeldt, Bram van Dijk, Annemarie Boleij, Martijn A. Huynen, and Bas E. Dutilh (2022), "Nutrition or nature: using elementary flux modes to disentangle the complex forces shaping prokaryote pan-genomes", BMC Ecology and Evolution 22: 101, doi: 10.1186/s12862-022-02052-3. Pubmed.
Background: Microbial pan-genomes are shaped by a complex combination of stochastic and deterministic forces. Even closely related genomes exhibit extensive variation in their gene content. Understanding what drives this variation requires exploring the interactions of gene products with each other and with the organism's external environment. However, to date, conceptual models of pan-genome dynamics often represent genes as independent units and provide limited information about their mechanistic interactions. Results: We simulated the stochastic process of gene-loss using the pooled genome-scale metabolic reaction networks of 46 taxonomically diverse bacterial and archaeal families as proxies for their pan-genomes. The frequency by which reactions are retained in functional networks when stochastic gene loss is simulated in diverse environments allowed us to disentangle the metabolic reactions whose presence depends on the metabolite composition of the external environment (constrained by "nutrition") from those that are independent of the environment (constrained by "nature"). By comparing the frequency of reactions from the first group with their observed frequencies in bacterial and archaeal families, we predicted the metabolic niches that shaped the genomic composition of these lineages. Moreover, we found that the lineages that were shaped by a more diverse metabolic niche also occur in more diverse biomes as assessed by global environmental sequencing datasets. Conclusion: We introduce a computational framework for analyzing and interpreting pan-reactomes that provides novel insights into the ecological and evolutionary drivers of pan-genome dynamics.
Ernestina Hauptfeld, Jordi Pelkmans, Terry T. Huisman, Armin Anocic, Basten Snoek, Bastiaan von Meijenfeldt, Jan Gerritse, Johan van Leeuwen, Gert Leurink, Arie van Lit, Ruud van Uffelen, Margot C. Koster, and Bas E. Dutilh (2022), "A metagenomic portrait of the microbial community responsible for two decades of bioremediation of poly-contaminated groundwater", Water Research 221: 118767, doi: 10.1016/j.watres.2022.118767. Pubmed. UU News item, interview with Tina on BNR radio.
|
Biodegradation of pollutants is a sustainable and cost-effective solution to groundwater pollution. Here, we investigate microbial populations involved in biodegradation of poly-contaminants in a pipeline for heavily contaminated groundwater. Groundwater moves from a polluted park to a treatment plant, where an aerated bioreactor effectively removes the contaminants. While the biomass does not settle in the reactor, sediment is collected afterwards and used to seed the new polluted groundwater via a backwash cycle. The pipeline has successfully operated since 1999, but the biological components in the reactor and the contaminated park groundwater have never been described. We sampled seven points along the pipeline, representing the entire remediation process, and characterized the changing microbial communities using genome-resolved metagenomic analysis. We assembled 297 medium- and high-quality metagenome-assembled genome sequences representing on average 46.3% of the total DNA per sample. We found that the communities cluster into two distinct groups, separating the anaerobic communities in the park groundwater from the aerobic communities inside the plant. In the park, the community is dominated by members of the genus Sulfuricurvum, while the plant is dominated by generalists from the order Burkholderiales. Known aromatic compound biodegradation pathways are four times more abundant in the plant-side communities compared to the park-side. Our findings provide a genome-resolved portrait of the microbial community in a highly effective groundwater treatment system that has treated groundwater with a complex contamination profile for two decades. |

|
Patrick A. de Jonge, Koen Wortelboer, Torsten P.M. Scheithauer, Bert-Jan H. van den Born, Aeilko H. Zwinderman, Franklin L. Nobrega, Bas E. Dutilh, Max Nieuwdorp, and Hilde Herrema (2022), "Gut virome profiling identifies a widespread bacteriophage family associated with metabolic syndrome", Nature Communications 13: 3594, doi: 10.1038/s41467-022-31390-5. Pubmed.
There is significant interest in altering the course of cardiometabolic disease development via gut microbiomes. Nevertheless, the highly abundant phage members of the complex gut ecosystem -which impact gut bacteria- remain understudied. Here, we show gut virome changes associated with metabolic syndrome (MetS), a highly prevalent clinical condition preceding cardiometabolic disease, in 196 participants by combined sequencing of bulk whole genome and virus like particle communities. MetS gut viromes exhibit decreased richness and diversity. They are enriched in phages infecting Streptococcaceae and Bacteroidaceae and depleted in those infecting Bifidobacteriaceae. Differential abundance analysis identifies eighteen viral clusters (VCs) as significantly associated with either MetS or healthy viromes. Among these are a MetS-associated Roseburia VC that is related to healthy control-associated Faecalibacterium and Oscillibacter VCs. Further analysis of these VCs revealed the Candidatus Heliusviridae, a highly widespread gut phage lineage found in 90+% of participants. The identification of the temperate Ca. Heliusviridae provides a starting point to studies of phage effects on gut bacteria and the role that this plays in MetS.
Sean Meaden, Ambarish Biswas, Ksenia Arkhipova, Sergio E. Morales, Bas E. Dutilh, Edze R. Westra, and Peter C. Fineran (2022), "High viral abundance and low diversity are associated with increased CRISPR-Cas prevalence across microbial ecosystems", Current Biology 32: 220-227.e5, doi: 10.1016/j.cub.2021.10.038. Pubmed.
CRISPR-Cas are adaptive immune systems that protect their hosts against viruses and other parasitic mobile genetic elements. Although widely distributed among prokaryotic taxa, CRISPR-Cas systems are not ubiquitous. Like most defense-system genes, CRISPR-Cas are frequently lost and gained, suggesting advantages are specific to particular environmental conditions. Selection from viruses is assumed to drive the acquisition and maintenance of these immune systems in nature, and both theory and experiments have identified phage density and diversity as key fitness determinants. However, these approaches lack the biological complexity inherent in nature. Here, we exploit metagenomic data from 324 samples across diverse ecosystems to analyze CRISPR abundance in natural environments. For each metagenome, we quantified viral abundance and diversity to test whether these contribute to CRISPR-Cas abundance across ecosystems. We find a strong positive association between CRISPR-Cas abundance and viral abundance. In addition, when controlling for differences in viral abundance, CRISPR-Cas systems are more abundant when viral diversity is low, suggesting that such adaptive immune systems may offer limited protection when required to target a diverse viral community. CRISPR-Cas abundance also differed among environments, with environmental classification explaining roughly a quarter of the variation in CRISPR-Cas relative abundance. The relationships between CRISPR-Cas abundance, viral abundance, and viral diversity are broadly consistent across environments, providing robust evidence from natural ecosystems that supports predictions of when CRISPR is beneficial. These results indicate that viral abundance and diversity are major ecological factors that drive the selection and maintenance of CRISPR-Cas in microbial ecosystems.
Francisco Murilo Zerbini, Stuart G. Siddell, Arcady R. Mushegian, Peter J. Walker, Elliot J. Lefkowitz, Evelien M. Adriaenssens, Poliane Alfenas-Zerbini, Bas E. Dutilh, María Laura García, Sandra Junglen, Mart Krupovic, Jens H. Kuhn, Amy J. Lambert, Małgorzata Łobocka, Hanna M. Oksanen, David L. Robertson, Luisa Rubino, Sead Sabanadzovic, Peter Simmonds, Nobuhiro Suzuki, Koenraad Van Doorslaer, Anne-Mieke Vandamme, Arvind Varsani (2022), "Differentiating between viruses and virus species by writing their names correctly", Archives of Virology 167: 1231-1234, doi: 10.1007/s00705-021-05323-4, Pubmed.
Following the results of the International Committee on Taxonomy of Viruses (ICTV) Ratification Vote held in March 2021, a standard two-part "binomial nomenclature" is now the norm for naming virus species. Adoption of the new nomenclature is still in its infancy; thus, it is timely to reiterate the distinction between "virus" and "virus species" and to provide guidelines for naming and writing them correctly.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Karolina M. Andralojc, Mariano A. Molina, Mengjie Qiu, Bram Spruijtenburg, Menno Rasing, Bernard Pater, Martijn A. Huynen, Bas E. Dutilh, Thomas H.A. Ederveen, Duaa Elmelik, Albert G. Siebers, Diede Loopik, Ruud L.M. Bekkers, William P.J. Leenders, and Willem J.G. Melchers (2021), "Novel high-resolution targeted sequencing of the cervicovaginal microbiome", BMC Biology 19: 267, doi: https://doi.org/10.1186/s12915-021-01204-z, Pubmed.
The cervicovaginal microbiome (CVM) plays a significant role in women's cervical health and disease. Microbial alterations at the species level and characteristic community state types (CST) have been associated with acquisition and persistence of high-risk human papillomavirus (hrHPV) infections that may result in progression of cervical lesions to malignancy. Current sequencing methods, especially most commonly used multiplex 16S rRNA gene sequencing, struggle to fully clarify these changes because they generally fail to provide sufficient taxonomic resolution to adequately perform species-level associative studies. To improve CVM species designation, we designed a novel sequencing tool targeting microbes at the species taxonomic rank and examined its potential for profiling the CVM.
Bas E. Dutilh, Arvind Varsani, Yigang Tong, Peter Simmonds, Sead Sabanadzovic, Luisa Rubino, Simon Roux, Alejandro Reyes Muñoz, Cédric Lood, Elliot J. Lefkowitz, Jens H. Kuhn, Mart Krupovic, Robert A. Edwards, J. Rodney Brister, Evelien M. Adriaenssens, and Matthew B. Sullivan (2021), "Perspective on taxonomic classification of uncultivated viruses", Current Opinion in Virology 51: 207-215, doi: 10.1016/j.coviro.2021.10.011. Pubmed.
|
Historically, virus taxonomy has been limited to describing viruses that were readily cultivated in the laboratory or emerging in natural biomes. Metagenomic analyses, single-particle sequencing, and database mining efforts have yielded new sequence data on an astounding number of previously unknown viruses. As metagenomes are relatively free of biases, these data provide an unprecedented insight into the vastness of the virosphere, but to properly value the extent of this diversity it is critical that the viruses are taxonomically classified. Inclusion of uncultivated viruses has already improved the process as well as the understanding of the taxa, viruses, and their evolutionary relationships. The continuous development and testing of computational tools will be required to maintain a dynamic virus taxonomy that can accommodate the new discoveries. |

|
Daan M. van Vliet, F.A. Bastiaan von Meijenfeldt, Bas E. Dutilh, Laura Villanueva, Jaap S. Sinninghe Damsté, Alfons J.M. Stams, and Irene Sánchez-Andrea (2021), "The bacterial sulfur cycle in expanding dysoxic and euxinic marine waters", Environmental Microbiology 23: 2834-2857, doi: 10.1111/1462-2920.15265. Pubmed.
Dysoxic marine waters (DMW, <1 µM oxygen) are currently expanding in volume in the oceans, which has biogeochemical, ecological, and societal consequences on a global scale. In these environments, distinct bacteria drive an active sulfur cycle, which has only recently been recognized for open-ocean DMW. This review summarizes the current knowledge on these sulfur-cycling bacteria. Critical bottlenecks and questions for future research are specifically addressed. Sulfate-reducing bacteria (SRB) are core members of DMW. However, their roles are not entirely clear, and they remain largely uncultured. We found support for their remarkable diversity and taxonomic novelty by mining metagenome-assembled genomes from the Black Sea as model ecosystem. We highlight recent insights into the metabolism of key sulfur-oxidizing SUP05 and Sulfurimonas bacteria, and discuss the probable involvement of uncultivated SAR324 and BS-GSO2 bacteria in sulfur oxidation. Uncultivated Marinimicrobia bacteria with a presumed organoheterotrophic metabolism are abundant in DMW. Like SRB, they may use specific molybdoenzymes to conserve energy from the oxidation, reduction or disproportionation of sulfur cycle intermediates such as S0 and thiosulfate, produced from the oxidation of sulfide. However, this complex network of reactions is yet to be constrained quantitatively.
Nikolaos Pappas and Bas E. Dutilh (2021) "Finding functional associations between prokaryotic virus orthologous groups: a proof of concept", BMC Bioinformatics 22: 438, doi: 10.1186/s12859-021-04343-w. Pubmed.
Background: The field of viromics has greatly benefited from recent developments in metagenomics, with significant efforts focusing on viral discovery. However, functional annotation of the increasing number of viral genomes is lagging behind. This is highlighted by the degree of annotation of the protein clusters in the prokaryotic Virus Orthologous Groups (pVOGs) database, with 83% of its current 9518 pVOGs having an unknown function. Results: In this study we describe a machine learning approach to explore potential functional associations between pVOGs. We measure seven genomic features and use them as input to a Random Forest classifier to predict protein-protein interactions between pairs of pVOGs. After systematic evaluation of the model's performance on 10 different datasets, we obtained a predictor with a mean accuracy of 0.77 and Area Under Receiving Operation Characteristic (AUROC) score of 0.83. Its application to a set of 2,133,027 pVOG-pVOG interactions allowed us to predict 267,265 putative interactions with a reported probability greater than 0.65. At an expected false discovery rate of 0.27, we placed 95.6% of the previously unannotated pVOGs in a functional context, by predicting their interaction with a pVOG that is functionally annotated. Conclusions: We believe that this proof-of-concept methodology, wrapped in a reproducible and automated workflow, can represent a significant step towards obtaining a more complete picture of bacteriophage biology.
Selma L. van Esveld, Lisette Meerstein-Kessel, Cas Boshoven, Jochem F. Baaij, Konstantin Barylyuk, Jordy P.M. Coolen, Joeri van Strien, Ronald A.J. Duim, Bas E. Dutilh, Daniel R. Garza, Marijn Letterie, Nicholas I. Proellochs, Michelle N. de Ridder, Prashanna Balaji Venkatasubramanian, Laura E. de Vries, Ross F. Waller, Taco W. A. Kooij, and Martijn A. Huynen (2021) "A prioritized and validated resource of mitochondrial proteins in Plasmodium identifies unique biology", mSphere 6: e0061421, doi: 10.1128/mSphere.00614-21. Pubmed.
Plasmodium species have a single mitochondrion that is essential for their survival and has been successfully targeted by antimalarial drugs. Most mitochondrial proteins are imported into this organelle, and our picture of the Plasmodium mitochondrial proteome remains incomplete. Many data sources contain information about mitochondrial localization, including proteome and gene expression profiles, orthology to mitochondrial proteins from other species, coevolutionary relationships, and amino acid sequences, each with different coverage and reliability. To obtain a comprehensive, prioritized list of Plasmodium falciparum mitochondrial proteins, we rigorously analyzed and integrated eight data sets using Bayesian statistics into a predictive score per protein for mitochondrial localization. At a corrected false discovery rate of 25%, we identified 445 proteins with a sensitivity of 87% and a specificity of 97%. They include proteins that have not been identified as mitochondrial in other eukaryotes but have characterized homologs in bacteria that are involved in metabolism or translation. Mitochondrial localization of seven Plasmodium berghei orthologs was confirmed by epitope labeling and colocalization with a mitochondrial marker protein. One of these belongs to a newly identified apicomplexan mitochondrial protein family that in P. falciparum has four members. With the experimentally validated mitochondrial proteins and the complete ranked P. falciparum proteome, which we have named PlasmoMitoCarta, we present a resource to study unique proteins of Plasmodium mitochondria.
Mart Krupovic, Dann Turner, Vera Morozova, Mike Dyall-Smith, Hanna M. Oksanen, Rob Edwards, Bas E. Dutilh, Susan M. Lehman, Alejandro Reyes, Diana P. Baquero, Matthew B. Sullivan, Jumpei Uchiyama, Jesca Nakavuma, Jakub Barylski, Mark J. Young, Shishen Du, Poliane Alfenas-Zerbini, Alla Kushkina, Andrew M. Kropinski, Ipek Kurtböke, J. Rodney Brister, Cédric Lood, B. L. Sarkar, Tong Yigang, Ying Liu, Li Huang, Johannes Wittmann, Nina Chanishvili, Leonardo J. van Zyl, Janis Rumnieks, Tomohiro Mochizuki, Matti Jalasvuori, Ramy K. Aziz, Małgorzata Łobocka, Kenneth M. Stedman, Andrey N. Shkoporov, Annika Gillis, Xu Peng, François Enault, Petar Knezevic, Rob Lavigne, Sung-Keun Rhee, Virginija Cvirkaite-Krupovic, Cristina Moraru, Andrea I. Moreno Switt, Minna M. Poranen, Andrew Millard, David Prangishvili, and Evelien M. Adriaenssens (2021) "Bacterial Viruses Subcommittee and Archaeal Viruses Subcommittee of the ICTV: update of taxonomy changes in 2021", Archives of Virology 166: 3239-3244, doi: 10.1007/s00705-021-05205-9. Pubmed.
In this article, we - the Bacterial Viruses Subcommittee and the Archaeal Viruses Subcommittee of the International Committee on Taxonomy of Viruses (ICTV) - summarise the results of our activities for the period March 2020 - March 2021. We report the division of the former Bacterial and Archaeal Viruses Subcommittee in two separate Subcommittees, welcome new members, a new Subcommittee Chair and Vice Chair, and give an overview of the new taxa that were proposed in 2020, approved by the Executive Committee and ratified by vote in 2021. In particular, a new realm, three orders, 15 families, 31 subfamilies, 734 genera and 1845 species were newly created or redefined (moved/promoted).
Stijn P. Andeweg, Can Keşmir, and Bas E. Dutilh (2020), "Quantifying the impact of Human Leukocyte Antigen on the human gut microbiota", mSphere 6: e00476-21 (preprint), Pubmed, doi: 10.1128/mSphere.00476-21.
The composition of the gut microbiota is affected by a number of factors, including the innate and adaptive immune system. The major histocompatibility complex (MHC), or the human leukocyte antigen (HLA) in humans, performs an essential role in vertebrate immunity and is very polymorphic in different populations. HLA determines the specificity of T lymphocyte and natural killer (NK) cell responses, including those against the commensal bacteria present in the human gut. Thus, it is likely that our HLA molecules, and thereby the adaptive immune response, can shape the composition of our microbiota. Here, we investigated the effect of HLA haplotype on the microbiota composition. We performed HLA typing and microbiota composition analyses on 3,002 public human gut microbiome data sets. We found that individuals with functionally similar HLA molecules are also similar in their microbiota composition. Our results show a statistical association between host HLA haplotype and gut microbiota composition. Because the HLA haplotype is a readily measurable parameter of the human immune system, these results open the door to incorporating the genetics of the immune system into predictive microbiome models.
Peter J. Walker, Stuart G. Siddell, Elliot J. Lefkowitz, Arcady R. Mushegian, Evelien M. Adriaenssens, Poliane Alfenas-Zerbini, Andrew J. Davison, Donald M. Dempsey, Bas E. Dutilh, Małgorzata Łobocka, Max L. Nibert, Hanna M. Oksanen, Richard J. Orton, David L. Robertson, Luisa Rubino, Sead Sabanadzovic, Peter Simmonds, Donald B. Smith, Nobuhiro Suzuki, Koenraad Van Dooerslaer, Anne-Mieke Vandamme, Arvind Varsani, Francisco Murilo Zerbini (2021), "Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021)", Archives of Virology 166: 2633-2648, doi: 10.1007/s00705-021-05156-1, Pubmed.
This article reports the changes to virus taxonomy approved and ratified by the International Committee on Taxonomy of Viruses (ICTV) in March 2021. The entire ICTV was invited to vote on 290 taxonomic proposals approved by the ICTV Executive Committee at its meeting in October 2020, as well as on the proposed revision of the International Code of Virus Classification and Nomenclature (ICVCN). All proposals and the revision were ratified by an absolute majority of the ICTV members. Of note, ICTV mandated a uniform rule for virus species naming, which will follow the binomial 'genus-species' format with or without Latinized species epithets. The Study Groups are requested to convert all previously established species names to the new format. ICTV has also abolished the notion of a type species, i.e., a species chosen to serve as a name-bearing type of a virus genus. The remit of ICTV has been clarified through an official definition of 'virus' and several other types of mobile genetic elements. The ICVCN and ICTV Statutes have been amended to reflect these changes.
Carlijn E. Bruggeling, Daniel R. Garza, Soumia Achouiti, Wouter Mes, Bas E. Dutilh, and Annemarie Boleij (2021) "Optimized bacterial DNA isolation method for microbiome analysis of human tissues", Microbiology Open 10: e1191, doi: 10.1002/mbo3.1191. Pubmed.
Recent advances in microbiome sequencing have rendered new insights into the role of the microbiome in human health with potential clinical implications. Unfortunately, the presence of host DNA in tissue isolates has hampered the analysis of host-associated bacteria. Here, we present a DNA isolation protocol for tissue, optimized on biopsies from resected human colons (~2-5 mm in size), which includes reduction of human DNA without distortion of relative bacterial abundance at the phylum level. We evaluated which concentrations of Triton and saponin lyse human cells and leave bacterial cells intact, in combination with DNAse treatment to deplete released human DNA. Saponin at a concentration of 0.0125% in PBS lysed host cells, resulting in a 4.5-fold enrichment of bacterial DNA while preserving the relative abundance of Firmicutes, Bacteroidetes, Gammaproteobacteria, and Actinobacteria assessed by qPCR. Our optimized protocol was validated in the setting of two large clinical studies on 521 in vivo acquired colon biopsies of 226 patients using shotgun metagenomics. The resulting bacterial profiles exhibited alpha and beta diversities that are similar to the diversities found by 16S rRNA amplicon sequencing. A direct comparison between shotgun metagenomics and 16S rRNA amplicon sequencing of 15 forceps tissue biopsies showed similar bacterial profiles and a similar Shannon diversity index between the sequencing methods. Hereby, we present the first protocol for enriching bacterial DNA from tissue biopsies that allows efficient isolation of all bacteria. Our protocol facilitates analysis of a wide spectrum of bacteria of clinical tissue samples improving their applicability for microbiome research.
Felipe H. Coutinho, Asier Zaragoza,Solas, Mario López-Pérez, Jakub Barylski, Andrzej Zielezinski, Bas E. Dutilh, Robert Edwards, and Francisco Rodriguez-Valera (2021), "RaFAH: Host prediction for viruses of Bacteria and Archaea based on protein content", Patterns 2: 100274, doi: 10.1016/j.patter.2021.100274. Pubmed.
Culture-independent approaches have recently shed light on the genomic diversity of viruses of prokaryotes. One fundamental question when trying to understand their ecological roles is: which host do they infect? To tackle this issue we developed a machine-learning approach named Random Forest Assignment of Hosts (RaFAH), that uses scores to 43,644 protein clusters to assign hosts to complete or fragmented genomes of viruses of Archaea and Bacteria. RaFAH displayed performance comparable with that of other methods for virus-host prediction in three different benchmarks encompassing viruses from RefSeq, single amplified genomes, and metagenomes. RaFAH was applied to assembled metagenomic datasets of uncultured viruses from eight different biomes of medical, biotechnological, and environmental relevance. Our analyses led to the identification of 537 sequences of archaeal viruses representing unknown lineages, whose genomes encode novel auxiliary metabolic genes, shedding light on how these viruses interfere with the host molecular machinery. RaFAH is available at https://sourceforge.net/projects/rafah/.
Michael Sheinman, Ksenia Arkhipova, Peter F. Arndt, Bas E. Dutilh, Rutger Hermsen, and Florian Massip (2021), "Identical sequences found in distant genomes reveal frequent horizontal transfer across the bacterial domain", eLife 10: e62719, Pubmed, doi: 10.7554/eLife.62719.
Horizontal Gene Transfer (HGT) is an essential force in microbial evolution. Despite detailed studies on a variety of systems, a global picture of HGT in the microbial world is still missing. Here, we exploit that HGT creates long identical DNA sequences in the genomes of distant species, which can be found efficiently using alignment-free methods. Our pairwise analysis of 93 481 bacterial genomes identified 138 273 HGT events. We developed a model to explain their statistical properties as well as estimate the transfer rate between pairs of taxa. This reveals that long-distance HGT is frequent: our results indicate that HGT between species from different phyla has occurred in at least 8% of the species. Finally, our results confirm that the function of sequences strongly impacts their transfer rate, which varies by more than 3 orders of magnitude between different functional categories. Overall, we provide a comprehensive view of HGT, illuminating a fundamental process driving bacterial evolution.
Katherine R. Amato, Marie-Clare Arrieta, Meghan B. Azad, Michael T. Bailey, Josiane L. Broussard, Carlijn E. Bruggeling, Erika C. Claud, Elizabeth K Costello, Emily R. Davenport, Bas E. Dutilh, Holly A. Swain Ewald, Paul Ewald, Erin C. Hanlon, Wrenetha Julion, Ali Keshavarzian, Corinne F. Maurice, Gregory E. Miller, Geoffrey A. Preidis, Laure Ségurel, Burton Singer, Sathish Subramanian, Liping Zhao, and Christopher W. Kuzawa (2021), "The human gut microbiome and health inequities", PNAS 118: e2017947118, doi: 10.1073/pnas.2017947118. Pubmed. EVMI, MedicalXpress, MedicalFacts, UA, Radboudumc (in Dutch), Sciencenewsnet, Sciencenewsnet, UU, Zorgkrant.
Individuals who are minoritized as a result of race, sexual identity, gender, or socioeconomic status experience a higher prevalence of many diseases. Understanding the biological processes that cause and maintain these socially driven health inequities is essential for addressing them. The gut microbiome is strongly shaped by host environments and affects host metabolic, immune, and neuroendocrine functions, making it an important pathway by which differences in experiences caused by social, political, and economic forces could contribute to health inequities. Nevertheless, few studies have directly integrated the gut microbiome into investigations of health inequities. Here, we argue that accounting for host-gut microbe interactions will improve understanding and management of health inequities, and that health policy must begin to consider the microbiome as an important pathway linking environments to population health.
|
The human gut microbiome consists of bacteria, archaea, eukaryotes, and viruses. The gut viruses are relatively underexplored. Here, we longitudinally analyzed the gut virome composition in 11 healthy adults: its stability, variation, and the effect of a gluten-free diet. Using viral enrichment and a de novo assembly-based approach, we demonstrate the quantitative dynamics of the gut virome, including dsDNA, ssDNA, dsRNA, and ssRNA viruses. We observe highly divergent individual viral communities, carrying on an average 2,143 viral genomes, 13.1% of which were present at all 3 time points. In contrast to previous reports, the Siphoviridae family dominates over Microviridae in studied individual viromes. We also show individual viromes to be stable at the family level but to vary substantially at the genera and species levels. Finally, we demonstrate that lower initial diversity of the human gut virome leads to a more pronounced effect of the dietary intervention on its composition. |

|
Winfried Goettsch, Niko Beerenwinkel, Li Deng, Lars Dölken, Bas E. Dutilh, Florian Erhard, Lars Kaderali, Max von Kleist, Roland Marquet, Jelle Matthijnssens, Shawna McCallin, Dino McMahon, Thomas Rattei, Ronald P. van Rij, David L. Robertson, Martin Schwemmle, Noam Stern-Ginossar, and Manja Marz (2021) "ITN-VIROINF: Understanding (harmful) virus-host interactions by linking virology and bioinformatics", Viruses 13: 766, doi: 10.3390/v13050766. Pubmed.
Many recent studies highlight the fundamental importance of viruses. Besides their important role as human and animal pathogens, their beneficial, commensal or harmful functions are poorly understood. By developing and applying tailored bioinformatical tools in important virological models, the Marie Skłodowska-Curie Initiative International Training Network VIROINF will provide a better understanding of viruses and the interaction with their hosts. This will open the door to validate methods of improving viral growth, morphogenesis and development, as well as to control strategies against unwanted microorganisms. The key feature of VIROINF is its interdisciplinary nature, which brings together virologists and bioinformaticians to achieve common goals.
Rahwa Taddese, Clara Belzer, Steven Aalvink, Marien I. de Jonge, Iris D. Nagtegaal, Bas E. Dutilh, and Annemarie Boleij (2021) "Production of inactivated gram-positive and gram-negative species with preserved cellular morphology and integrity", Journal of Microbiological Methods 184: 106208, doi: 10.1016/j.mimet.2021.106208. Pubmed.
There are many approaches available to produce inactive bacteria by termination of growth, each with a different efficacy, impact on cell integrity, and potential for application in standardized inactivation protocols. The aim of this study was to compare these approaches and develop a standardized protocol for generation of inactivated Gram-positive and Gram-negative bacteria, yielding cells that are metabolically dead with retained cellular integrity i.e., preserving the surface and limited leakage of intracellular proteins and DNA. These inactivated bacteria are required for various applications, for instance, when investigating receptor-triggered signaling or bacterial contact-dependent analysis of cell lines requiring long incubation times. We inactivated eight different bacterial strains of different species by treatment with beta-propiolactone, ethanol, formalin, sodium hydroxide, and pasteurization. Inactivation efficacy was determined by culturing, and cell wall integrity assessed by quantifying released DNA, bacterial membrane and intracellular DNA staining, and visualization by scanning electron microscopy. Based on these results, we discuss the bacterial inactivation methods, and their advantages and disadvantages to study host-microbe interactions with inactivated bacteria.
Martin T. Jahn, Tim Lachnit, Sebastian M. Markert, Christian Stigloher, Lucia Pita, Marta Ribes, Bas E. Dutilh, and Ute Hentschel (2021), "Lifestyle of sponge symbiont phages by host prediction and correlative microscopy", ISME Journal 15: 2001-2011, doi: 10.1038/s41396-021-00900-6. Pubmed. Behind the paper.
Bacteriophages (phages) are ubiquitous elements in nature, but their ecology and role in animals remains little understood. Sponges represent the oldest known extant animal-microbe symbiosis and are associated with dense and diverse microbial consortia. Here we investigate the tripartite interaction between phages, bacterial symbionts, and the sponge host. We combined imaging and bioinformatics to tackle important questions on who the phage hosts are and what the replication mode and spatial distribution within the animal is. This approach led to the discovery of distinct phage-microbe infection networks in sponge versus seawater microbiomes. A new correlative in situ imaging approach ('PhageFISH-CLEM') localised phages within bacterial symbiont cells, but also within phagocytotically active sponge cells. We postulate that the phagocytosis of free virions by sponge cells modulates phage-bacteria ratios and ultimately controls infection dynamics. Prediction of phage replication strategies indicated a distinct pattern, where lysogeny dominates the sponge microbiome, likely fostered by sponge host-mediated virion clearance, while lysis dominates in seawater. Collectively, this work provides new insights into phage ecology within sponges, highlighting the importance of tripartite animal-phage-bacterium interplay in holobiont functioning. We anticipate that our imaging approach will be instrumental to further understanding of viral distribution and cellular association in animal hosts.
Felipe H. Coutinho, F.A. Bastiaan von Meijenfeldt, Juline M. Walter, Jose M. Haro-Moreno, Mario Lopéz-Pérez, Marcel C. van Verk, Cristiane C. Thompson, Carlos A.N. Cosenza, Luciana Appolinario, Rodolfo Paranhos, Anderson Cabral, Bas E. Dutilh, and Fabiano L. Thompson (2021), "Ecogenomics and metabolic potential of the South Atlantic Ocean microbiome", Science of The Total Environment 765: 142758, doi: 10.1016/j.scitotenv.2020.142758. Pubmed.
The unique combination of depth, salinity, and water masses make the South Atlantic Ocean an ecosystem of special relevance within the global ocean. Yet, the microbiome of this ecosystem has received less attention than other regions of the global Ocean. This has hampered our understanding of the diversity and metabolic potential of the microorganisms that dwell in this habitat. To fill this knowledge gap, we analyzed a collection of 31 metagenomes from the Atlantic Ocean that spanned the epipelagic, mesopelagic and bathypelagic zones (surface to 4000 m). Read-centric and gene-centric analysis revealed the unique taxonomic and functional composition of metagenomes from each depth zone, which was driven by differences in physical and chemical parameters. In parallel, a total of 40 metagenome-assembled genomes were obtained, which recovered one third of the total community. Phylogenomic reconstruction revealed that many of these genomes are derived from poorly characterized taxa of Bacteria and Archaea. Genomes derived from heterotrophic bacteria of the aphotic zone displayed a large apparatus of genes suited for the utilization of recalcitrant organic compounds such as cellulose, chitin and alkanes. In addition, we found genomic evidence suggesting that mixotrophic bacteria from the bathypelagic zone could perform carbon fixation through the Calvin-Benson-Bassham cycle, fueled by sulfur oxidation. Finally, we found that the viral communities shifted throughout the water column regarding their targeted hosts and virus-to-microbe ratio, in response to shifts in the composition and functioning their microbial counterparts. Our findings shed light on the microbial and viral drivers of important biogeochemical processes that take place in the South Atlantic Ocean.
Laura Villanueva*, F.A. Bastiaan von Meijenfeldt*, Alexander B. Westbye, Subhash Yadav, Ellen C. Hopmans, Bas E. Dutilh†, and Jaap S. Sinninghe Damsté† (2021), "Bridging the membrane lipid divide: bacteria of the FCB group superphylum have the potential to synthesize archaeal ether lipids", ISME Journal 15: 168-182, doi: 10.1038/s41396-020-00772-2, Pubmed. *,†Equal contributions. News: NRC.
|
Archaea synthesize membranes of isoprenoid lipids that are ether-linked to glycerol-1-phosphate (G1P), while Bacteria/Eukarya produce membranes consisting of fatty acids ester-bound to glycerol-3-phosphate (G3P). This dichotomy in membrane lipid composition (i.e., the 'lipid divide') is believed to have arisen after the Last Universal Common Ancestor (LUCA). A leading hypothesis is that LUCA possessed a heterochiral 'mixed archaeal/bacterial membrane'. However, no natural microbial representatives supporting this scenario have been shown to exist today. Here, we demonstrate that bacteria of the Fibrobacteres-Chlorobi-Bacteroidetes (FCB) group superphylum encode a putative archaeal pathway for ether-bound isoprenoid membrane lipids in addition to the bacterial fatty acid membrane pathway. Key genes were expressed in the environment and their recombinant expression in Escherichia coli resulted in the formation of a 'mixed archaeal/bacterial membrane'. Genomic evidence and biochemical assays suggest that the archaeal-like lipids of members of the FCB group could possess either a G1P or G3P stereochemistry. Our results support the existence of 'mixed membranes' in natural environments and their stability over a long period in evolutionary history, thereby bridging a once-thought fundamental divide in biology. |
|
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
João C. Setubal, Jens Stoye, and Bas E. Dutilh (2020), "Editorial: Computational methods for microbiome analysis", Frontiers in Genetics 11: 623897, Pubmed, doi: 10.3389/fgene.2020.623897. Find the Research Topic and associated E-book here: Computational Methods for Microbiome Analysis.
Microbes play critical roles in the lives of hosts (plants, animals, humans) and in almost any environment one can think of. Gathering microbiome sequence data has become easier and cheaper than ever before, leading to an exponential growth in the amount of such data available for analysis. With this explosion has come a pressing need for sophisticated computational tools that can help make sense of these datasets. Current challenges, such as the complexity of microbiome-host-environment interactions and the large sizes of datasets, make for a fascinating research field. The goal of this Research Topic was to gather a collection of high-quality original papers on the general theme of computational methods for microbiome analysis. We now present the results, which consist of 13 papers.
Franklin L. Nobrega, Hielke Walinga, Bas E. Dutilh, and Stan J.J. Brouns (2020), "Prophages are associated with extensive CRISPR-Cas auto-immunity", Nucleic Acids Research 48: 12074-12084, (preprint), Pubmed, doi: 10.1093/nar/gkaa1071.
CRISPR-Cas systems require discriminating self from non-self DNA during adaptation and interference. Yet, multiple cases have been reported of bacteria containing self-targeting spacers (STS), i.e. CRISPR spacers targeting protospacers on the same genome. STS has been suggested to reflect potential auto-immunity as an unwanted side effect of CRISPR-Cas defense, or a regulatory mechanism for gene expression. Here we investigated the incidence, distribution, and evasion of STS in over 100 000 bacterial genomes. We found STS in all CRISPR-Cas types and in one fifth of all CRISPR-carrying bacteria. Notably, up to 40% of I-B and I-F CRISPR-Cas systems contained STS. We observed that STS-containing genomes almost always carry a prophage and that STS map to prophage regions in more than half of the cases. Despite carrying STS, genetic deterioration of CRISPR-Cas systems appears to be rare, suggesting a level of escape from the potentially deleterious effects of STS by other mechanisms such as anti-CRISPR proteins and CRISPR target mutations. We propose a scenario where it is common to acquire an STS against a prophage, and this may trigger more extensive STS buildup by primed spacer acquisition in type I systems, without detrimental autoimmunity effects as mechanisms of auto-immunity evasion create tolerance to STS-targeted prophages.
Nikolaos Pappas, Simon Roux, Martin Hölzer, Kevin Lamkiewicz, Florian Mock, Manja Marz, and Bas E. Dutilh (2020), "Virus bioinformatics", Reference Module in Life Sciences 1: 124-132, doi: 10.1016/B978-0-12-814515-9.00034-5. Pubmed.
Since the discovery of computers, bioinformatics and computational biology have been instrumental in a wide range of discoveries in virology. These include early mathematical models of virus-host interaction, and more recently the analysis of viral nucleotide and protein sequences to track their function, epidemiology, and evolution. The genomics revolution has provided an unprecedented amount of sequence information from both viruses and their hosts. In this article, we discuss how bioinformatics allows viral sequence data to be analyzed and interpreted, including an overview of commonly used tools and examples of applications.
Isabel Douterelo, Bas E. Dutilh, Carolina Calero, E. Rosales, and Stewart Husband (2020), "Impact of phosphate dosing on the microbial ecology of drinking water distribution systems: fieldwork studies in chlorinated networks", Water Research 187: 116416, Pubmed, doi: 10.1016/j.watres.2020.116416.
|
Phosphate is routinely dosed to ensure regulatory compliance for lead in drinking water distribution systems. Little is known about the impact of the phosphate dose on the microbial ecology in these systems and in particular the endemic biofilms. Disturbance of the biofilms and embedded material in distribution can cause regulatory failures for turbidity and metals. To investigate the impact of phosphate on developing biofilms, pipe wall material from four independent pipe sections was mobilised and collected using two twin-flushing operations a year apart in a chlorinated UK network pre- and post-phosphate dosing. Intensive monitoring was undertaken, including turbidity and water physico-chemistry, traditional microbial culture-based indicators, and microbial community structure via sequencing the 16S rRNA gene for bacteria and the ITS2 gene for fungi. Whole metagenome sequencing was used to study shifts in functional characteristics following the addition of phosphate. |

|
| As an operational consequence, turbidity responses from the phosphate-enriched water were increased, particularly from cast iron pipes. Differences in the taxonomic composition of both bacteria and fungi were also observed, emphasising a community shift towards microorganisms able to use or metabolise phosphate. Phosphate increased the relative abundance of bacteria such as Pseudomonas, Paenibacillus, Massilia, Acinetobacter and the fungi Cadophora, Rhizophagus and Eupenicillium. Whole metagenome sequencing showed with phosphate a favouring of sequences related to Gram-negative bacterium type cell wall function, virions and thylakoids, but a reduction in the number of sequences associated to vitamin binding, methanogenesis and toxin biosynthesis. With current faecal indicator tests only providing risk detection in bulk water samples, this work improves understanding of how network changes effect microbial ecology and highlights the potential for new approaches to inform future monitoring or control strategies to protect drinking water quality. | |
Patrick A. de Jonge, Dieuwke J.C. Smit Sibinga, Oliver A. Boright, Ana Rita Costa, Franklin L. Nobrega, Stan J.J. Brouns, and Bas E. Dutilh (2020), "Development of styrene maleic acid lipid particles (SMALPs) as a tool for studies of phage-host interactions", Journal of Virology 94: e01559-20, doi: 10.1128/JVI.01559-20, Pubmed.
The infection of a bacterium by a phage starts with attachment to a receptor molecule on the host cell surface by the phage. As receptor-phage interactions are crucial to successful infections, they are major determinants of phage host-range and by extension of the broader effects that phages have on bacterial communities. Many receptor molecules, particularly membrane proteins, are difficult to isolate because their stability is supported by their native membrane environments. Styrene maleic acid lipid particles (SMALPs), a recent advance in membrane protein studies, are the result of membrane solubilizations by styrene maleic acid (SMA) co-polymer chains. SMALPs thereby allow for isolation of membrane proteins while maintaining their native environment. Here, we explore SMALPs as a tool to isolate and study phage-receptor interactions. We show that SMALPs produced from taxonomically distant bacterial membranes allow for receptor-specific decrease of viable phage counts of several model phages that span the three largest phage families. After characterizing the effects of incubation time and SMALP concentration on the activity of three distinct phages, we present evidence that the interaction between two model phages and SMALPs is specific to bacterial species and the phage receptor molecule. These interactions additionally lead to DNA ejection by nearly all particles at high phage titers. We conclude that SMALPs are a potentially highly useful tool for phage host-interaction studies.
Alessandro Rossi, Laura Treu, Stefano Toppo, Henrike Zschach, Stefano Campanaro, and Bas E. Dutilh (2020), "Evolutionary study of the crAssphage virus at gene level", Viruses 12: 1035, doi: 10.3390/v12091035, Pubmed.
crAss-like viruses are a putative family of bacteriophages recently discovered. The eponym of the clade, crAssphage, is an enteric bacteriophage estimated to be present in at least half of the human population and it constitutes up to 90% of the sequences in some human fecal viral metagenomic datasets. We focused on the evolutionary dynamics of the genes encoded on the crAssphage genome. By investigating the conservation of the genes, a consistent variation in the evolutionary rates across the different functional groups was found. Gene duplications in crAss-like genomes were detected. By exploring the differences among the functional categories of the genes, we confirmed that the genes encoding capsid proteins were the most ubiquitous, despite their overall low sequence conservation. It was possible to identify a core of proteins whose evolutionary trees strongly correlate with each other, suggesting their genetic interaction. This group includes the capsid proteins, which are thus established as extremely suitable for rebuilding the phylogenetic tree of this viral clade. A negative correlation between the ubiquity and the conservation of viral protein sequences was shown. Together, this study provides an in-depth picture of the evolution of different genes in crAss-like viruses.
Rahwa Taddese*, Daniel R. Garza*, Lilian N. Ruiter, Marien I. de Jonge, Clara Belzer, Steven Aalvink, Iris D. Nagtegaal, Bas E. Dutilh, and Annemarie Boleij (2020), "Growth rate alterations of human colorectal cancer cells by 157 gut bacteria", Gut Microbes 12: 1-20, doi: 10.1080/19490976.2020.1799733, Pubmed. *Equal contributions.
Several bacteria in the human gut microbiome have been associated with colorectal cancer (CRC) by high-throughput screens. In some cases, molecular mechanisms have been elucidated that drive tumorigenesis, including bacterial membrane proteins or secreted molecules that interact with the human cancer cells. For most gut bacteria, however, it remains unknown if they enhance or inhibit cancer cell growth. Here, we screened bacteria-free supernatants (secretomes) and inactivated cells of over 150 cultured bacterial strains for their effects on cell growth. We observed family-level and strain-level effects that often differed between bacterial cells and secretomes, suggesting that different molecular mechanisms are at play. Secretomes of Bacteroidaceae, Enterobacteriaceae, and Erysipelotrichaceae bacteria enhanced cell growth, while most Fusobacteriaceae cells and secretomes inhibited growth, contrasting prior findings. In some bacteria, the presence of specific functional genes was associated with cell growth rates, including the virulence genes TcdA, TcdB in Clostridiales and FadA in Fusobacteriaceae, which both inhibited growth. Bacteroidaceae cells that enhanced growth were enriched for genes of the cobalamin synthesis pathway, while Fusobacteriaceae cells that inhibit growth were enriched for genes of the ethanolamine utilization pathway. Together, our results reveal how different gut bacteria have wide-ranging effects on cell growth, contribute a better understanding of the effects of the gut microbiome on host cells, and provide a valuable resource for identifying candidate target genes for potential microbiome-based diagnostics and treatment strategies.
Marleen Balvert, Xiao Luo, Tina Hauptfeld, Alexander Schoenhuth, and Bas E. Dutilh (2019), "OGRE: Overlap Graph-based metagenomic Read clustEring", Bioinformatics 37: 905-912. doi: 10.1093/bioinformatics/btaa760, Pubmed.
Motivation: The microbes that live in an environment can be identified from the combined genomic material, also referred to as the metagenome. Sequencing a metagenome can result in large volumes of sequencing reads. A promising approach to reduce the size of metagenomic datasets is by clustering reads into groups based on their overlaps. Clustering reads is valuable to facilitate downstream analyses, including computationally intensive strain-aware assembly. As current read clustering approaches cannot handle the large datasets arising from high-throughput metagenome sequencing, a novel read clustering approach is needed. In this paper we propose OGRE, an Overlap Graph-based Read clustEring procedure for high-throughput sequencing data, with a focus on shotgun metagenomes. Results: We show that for small datasets OGRE outperforms other read binners in terms of the number of species included in a cluster, also referred to as cluster purity, and the fraction of all reads that is placed in one of the clusters. Furthermore, OGRE is able to process metagenomic datasets that are too large for other read binners into clusters with high cluster purity. Conclusion: OGRE is the only method that can successfully cluster reads in species-specific clusters for large metagenomic datasets without running into computation time- or memory issues. Availability: Code is made available on Github (https://github.com/Marleen1/OGRE).
Patrick A. de Jonge, F.A. Bastiaan von Meijenfeldt, Ana Rita Costa, Franklin L. Nobrega, Stan J.J. Brouns, and Bas E. Dutilh (2020), "Adsorption sequencing as a rapid method to link environmental bacteriophages to hosts", iScience 23: 101439, doi: 10.1016/j.isci.2020.101439, Pubmed.
|
An important viromics challenge is associating bacteriophages to hosts. To address this, we developed adsorption sequencing (AdsorpSeq), a readily implementable method to measure phages that are preferentially adsorbed to specific host cell envelopes. AdsorpSeq thus captures the key initial infection cycle step. Phages are added to cell envelopes, adsorbed phages are isolated through gel electrophoresis, after which adsorbed phage DNA is sequenced and compared with the full virome. Here, we show that AdsorpSeq allows for separation of phages based on receptor-adsorbing capabilities. Next, we applied AdsorpSeq to identify phages in a wastewater virome that adsorb to cell envelopes of nine bacteria, including important pathogens. We detected 26 adsorbed phages including common and rare members of the virome, a minority being related to previously characterized phages. We conclude that AdsorpSeq is an effective new tool for rapid characterization of environmental phage adsorption, with a proof-of-principle application to Gram-negative host cell envelopes. |

|
Peter J. Walker, Stuart G. Siddell, Elliot J. Lefkowitz, Arcady R. Mushegian, Evelien M. Adriaenssens, Donald M. Dempsey, Bas E. Dutilh, Balázs Harrach, Robert L. Harrison, R. Curtis Hendrickson, Sandra Junglen, Nick J. Knowles, Andrew M. Kropinski, Mart Krupovic, Jens H. Kuhn, Max Nibert, Richard J. Orton, Luisa Rubino, Sead Sabanadzovic, Peter Simmonds, Donald B. Smith, Arvind Varsani, Francisco Murilo Zerbini, and Andrew J. Davison (2020), "Changes to virus taxonomy and the Statutes ratified by the International Committee on Taxonomy of Viruses (2020)", Archives of Virology 165: 2737-2748, doi: 10.1007/s00705-020-04752-x, Pubmed.
This article reports the changes to virus classification and taxonomy approved and ratified by the International Committee on Taxonomy of Viruses (ICTV) in March 2020. The entire ICTV was invited to vote on 206 taxonomic proposals approved by the ICTV Executive Committee at its meeting in July 2019, as well as on the proposed revision of the ICTV Statutes. All proposals and the revision of the Statutes were approved by an absolute majority of the ICTV voting membership. Of note, ICTV has approved a proposal that extends the previously established realm Riboviria to encompass nearly all RNA viruses and reverse-transcribing viruses, and approved three separate proposals to establish three realms for viruses with DNA genomes.
Joël Klein, Manon Neilen, Marcel van Verk, Bas E. Dutilh, and Guido van den Ackerveken (2020), "Genome reconstruction of the non-culturable spinach downy mildew Peronospora effusa by metagenome filtering", PLoS ONE 15: e0225808, doi: 10.1371/journal.pone.0225808 Pubmed.
Peronospora effusa (previously known as P. farinosa f. sp. spinaciae, and here referred to as Pfs) is an obligate biotrophic oomycete that causes downy mildew on spinach (Spinacia oleracea). To combat this destructive many disease resistant cultivars have been bred and used. However, new Pfs races rapidly break the employed resistance genes. To get insight into the gene repertoire of Pfs and identify infection-related genes, the genome of the first reference race, Pfs1, was sequenced, assembled, and annotated. Due to the obligate biotrophic nature of this pathogen, material for DNA isolation can only be collected from infected spinach leaves that, however, also contain many other microorganisms. The obtained sequences can, therefore, be considered a metagenome. To filter and obtain Pfs sequences we utilized the CAT tool to taxonomically annotate ORFs residing on long sequences of a genome pre-assembly. This study is the first to show that CAT filtering performs well on eukaryotic contigs. Based on the taxonomy, determined on multiple ORFs, contaminating long sequences and corresponding reads were removed from the metagenome. Filtered reads were re-assembled to provide a clean and improved Pfs genome sequence of 32.4 Mbp consisting of 8,635 scaffolds. Transcript sequencing of a range of infection time points aided the prediction of a total of 13,277 gene models, including 99 RxLR(-like) effector, and 14 putative Crinkler genes. Comparative analysis identified common features in the predicted secretomes of different obligate biotrophic oomycetes, regardless of their phylogenetic distance. Their secretomes are generally smaller, compared to hemi-biotrophic and necrotrophic oomycete species. We observe a reduction in proteins involved in cell wall degradation, in Nep1-like proteins (NLPs), proteins with PAN/apple domains, and host translocated effectors. The genome of Pfs1 will be instrumental in studying downy mildew virulence and for understanding the molecular adaptations by which new isolates break spinach resistance.
Alexander E. Gorbalenya, Mart Krupovic, Arcady Mushegian, Andrew M. Kropinski, Stuart G. Siddell, Arvind Varsani, Michael J. Adams, Andrew J. Davison, Bas E. Dutilh, Balázs Harrach, Robert L. Harrison, Sandra Junglen, Andrew M.Q. King, Nick J. Knowles, Elliot J. Lefkowitz, Max L. Nibert, Luisa Rubino, Sead Sabanadzovic, Hélène Sanfaçon, Peter Simmonds, Peter J. Walker, F. Murilo Zerbini, and Jens H. Kuhn (2020), "The new scope of virus taxonomy: partitioning the virosphere into 15 hierarchical ranks", Nature Microbiology 5: 668-674, doi: 10.1038/s41564-020-0709-x. Pubmed. News at Utrecht University (in English and Dutch), Behind the paper.
Virus taxonomy emerged as a discipline in the middle of the twentieth century. Traditionally, classification by virus taxonomists has been focussed on the grouping of relatively closely related viruses. However, during the past few years, the International Committee on Taxonomy of Viruses (ICTV) has recognized that the taxonomy it develops can be usefully extended to include the basal evolutionary relationships among distantly related viruses. Consequently, the ICTV has changed its Code to allow a 15-rank classification hierarchy that closely aligns with the Linnaean taxonomic system and may accommodate the entire spectrum of genetic divergence in the virosphere. The current taxonomies of three human pathogens, Ebola virus, severe acute respiratory syndrome coronavirus and herpes simplex virus 1 are used to illustrate the impact of the expanded rank structure. This new rank hierarchy of virus taxonomy will stimulate further research on virus origins and evolution, and vice versa, and could promote crosstalk with the taxonomies of cellular organisms.
|
|

|
Evelien M. Adriaenssens, Matthew B. Sullivan, Petar Knezevic, Leonardo J. van Zyl, B.L. Sarkar, Bas E. Dutilh, Poliane Alfenas-Zerbini, Małgorzata Łobocka, Yigang Tong, James Rodney Brister, Andrea I. Moreno Switt, Jochen Klumpp, Ramy Karam Aziz, Jakub Barylski, Jumpei Uchiyama, Rob A. Edwards, Andrew M. Kropinski, Nicola K. Petty, Martha R.J. Clokie, Alla I. Kushkina, Vera V. Morozova, Siobain Duffy, Annika Gillis, Janis Rumnieks, İpek Kurtböke, Nina Chanishvili, Lawrence Goodridge, Johannes Wittmann, Rob Lavigne, Ho Bin Jang, David Prangishvili, Francois Enault, Dann Turner, Minna M. Poranen, Hanna M. Oksanen, and Mart Krupovic (2020), "Taxonomy of prokaryotic viruses: 2018-2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee", Archives of Virology 165: 1253-1260. Pubmed, doi: 10.1007/s00705-020-04577-8.
| Background Colorectal cancer (CRC) is a complex multifactorial disease. Increasing evidence suggests that the microbiome is involved in different stages of CRC initiation and progression. Beyond specific pro-oncogenic mechanisms found in pathogens, metagenomic studies indicate the existence of a microbiome signature, where particular bacterial taxa are enriched in the metagenomes of CRC patients. Here, we investigate to what extent the abundance of bacterial taxa in CRC metagenomes can be explained by the growth advantage resulting from the presence of specific CRC metabolites in the tumor microenvironment. Methods We composed lists of metabolites and bacteria that are enriched on CRC samples by reviewing metabolomics experimental literature and integrating data from metagenomic case-control studies. We computationally evaluated the growth effect of CRC enriched metabolites on over 1500 genome-based metabolic models of human microbiome bacteria. We integrated the metabolomics data and the mechanistic models by using scores that quantify the response of bacterial biomass production to CRC-enriched metabolites and used these scores to rank bacteria as potential CRC passengers. Results We found that metabolic networks of bacteria that are significantly enriched in CRC metagenomic samples either depend on metabolites that are more abundant in CRC samples or specifically benefit from these metabolites for biomass production. This suggests that metabolic alterations in the cancer environment are a major component shaping the CRC microbiome. Conclusion Here, we show with in sillico models that supplementing the intestinal environment with CRC metabolites specifically predicts the outgrowth of CRC-associated bacteria. We thus mechanistically explain why a range of CRC passenger bacteria are associated with CRC, enhancing our understanding of this disease. Our methods are applicable to other microbial communities, since it allows the systematic investigation of how shifts in the microbiome can be explained from changes in the metabolome. |
|
Juline M. Walter, Felipe H. Coutinho, Luciana Leomil, Paulo I. Hargreaves, Mariana E. Campeão, Verônica V. Vieira, Beatriz S. Silva, Giovana O. Fistarol, Paulo S. Salomon, Tomoo Sawabe, Sayaka Mino, Masashi Hosokawa, Hideaki Miyashita, Fumito Maruyama, Marcel C. van Verk, Bas E. Dutilh, Cristiane C. Thompson, and Fabiano L. Thompson (2020), "Ecogenomics of the marine benthic filamentous cyanobacterium Adonisia", Microbial Ecology 80: 249-265, Pubmed, doi: 10.1007/s00248-019-01480-x.
Turfs are among the major benthic components of reef systems worldwide. The nearly complete genome sequences, basic physiological characteristics, and phylogenomic reconstruction of two phycobiliprotein-rich filamentous cyanobacteria strains isolated from turf assemblages from the Abrolhos Bank (Brazil) are investigated. Both Adonisia turfae CCMR0081T (= CBAS 745T) and CCMR0082 contain approximately 8 Mbp in genome size and experiments identified that both strains exhibit chromatic acclimation. Whereas CCMR0081T exhibits chromatic acclimation type 3 (CA3) regulating both phycocyanin (PC) and phycoerythrin (PE), CCMR0082 strain exhibits chromatic acclimation type 2 (CA2), in correspondence with genes encoding specific photosensors and regulators for PC and PE. Furthermore, a high number and diversity of secondary metabolite synthesis gene clusters were identified in both genomes, and they were able to grow at high temperatures (28 °C, with scant growth at 30 °C). These characteristics provide insights into their widespread distribution in reef systems.
Isabel Douterelo, Bas E. Dutilh, Ksenia Arkhipova, Carolina Calero, and Stewart Husband (2020), "Microbial diversity, ecological networks and functional traits associated to materials used in drinking water distribution systems", Water Research 173: 115586, Pubmed, doi: 10.1016/j.watres.2020.115586.
Drinking water distribution systems host complex microbial communities as biofilms that interact continuously with delivered water. Understanding the diversity, behavioural and functional characteristics will be a requisite for developing future monitoring strategies and protection against water-borne health risks. To improve understanding, this study investigates mobilisation and accumulation behaviour, microbial community structure and functional variations of biofilms developing on different pipe materials from within an operational network. Samples were collected from four pipes during a repeated flushing operation three months after an initial visit that used hydraulic forces to mobilise regenerating biofilms yet without impacting the upstream network. To minimise confounding factors, test sections were chosen with comparable daily hydraulic regimes, physical dimensions, and all connected straight of a common trunk main and within close proximity, hence similar water chemistry, pressure and age. Taxonomical results showed differences in colonising communities between pipe materials, with several genera, including the bacteria Pseudomonas and the fungi Cladosporium, present in every sample. Diverse bacterial communities dominated compared to more homogeneous fungal, or mycobiome, community distribution. The analysis of bacterial/fungal networks based on relative abundance of operational taxonomic units (OTUs) indicated microbial communities from cast iron pipes were more stable than communities from the non-ferrous pipe materials. Novel analysis of functional traits between all samples were found to be mainly associated to mobile genetic elements that play roles in determining links between cells, including phages, prophages, transposable elements, and plasmids. The use of functional traits can be considered for development in future surveillance methods, capable of delivering network condition information beyond that of limited conventional faecal indicator tests, that will help protect water quality and public health.
Amber C.A. Hendriks, Frans A.G. Reubsaet, A.M.D. (Mirjam) Kooistra-Smid, John W.A. Rossen, Bas E. Dutilh, Aldert L. Zomer, and Maaike J.C. van den Beld on behalf of the IBESS group (2020), "Genome-wide association studies of Shigella spp. and Enteroinvasive Escherichia coli isolates demonstrate an absence of genetic markers for prediction of disease severity", BMC Genomics 21: 138, Pubmed, doi: 10.1186/s12864-020-6555-7.
Background We investigated the association of symptoms and disease severity of shigellosis patients with genetic determinants of infecting Shigella and entero-invasive Escherichia coli (EIEC), because determinants that predict disease outcome per individual patient could be used to prioritize control measures. For this purpose, genome wide association studies (GWAS) were performed using presence or absence of single genes, combinations of genes, and k-mers. All genetic variants were derived from draft genome sequences of isolates from a multicenter cross-sectional study conducted in the Netherlands during 2016 and 2017. Clinical data of patients consisting of binary/dichotomous representation of symptoms and their calculated severity scores were also available from this study. To verify the suitability of the methods used, the genetic differences between the genera Shigella and Escherichia were used as control. Results The isolates obtained were representative of the population structure encountered in other Western European countries. No association was found between single genes or combinations of genes and separate symptoms or disease severity scores. Our benchmark characteristic, genus, resulted in eight associated genes and >3,000,000 k-mers, indicating adequate performance of the algorithms used. Conclusions To conclude, using several microbial GWAS methods, genetic variants in Shigella spp. and EIEC that can predict specific symptoms or a more severe course of disease were not identified, suggesting that disease severity of shigellosis is dependent on other factors than the genetic variation of the infecting bacteria. Specific genes or gene fragments of isolates from patients are unsuitable to predict outcomes and cannot be used for development, prioritization and optimization of guidelines for control measures of shigellosis or infections with EIEC.
David Lähnemann, Johannes Köster, Ewa Szczurek, Davis J. McCarthy, Stephanie C. Hicks, Mark D. Robinson, Catalina A. Vallejos, Kieran R. Campbell, Niko Beerenwinkel, Ahmed Mahfouz, Luca Pinello, Pavel Skums, Alexandros Stamatakis, Camille Stephan-Otto Attolini, Samuel Aparicio, Jasmijn Baaijens, Marleen Balvert, Buys de Barbanson, Antonio Cappuccio, Giacomo Corleone, Bas E. Dutilh, Maria Florescu, Victor Guryev, Rens Holmer, Katharina Jahn, Thamar Jessurun Lobo, Emma M. Keizer, Indu Khatri, Szymon M. Kielbasa, Jan O. Korbel, Alexey M. Kozlov, Tzu-Hao Kuo, Boudewijn P.F. Lelieveldt, Ion I. Mandoiu, John C. Marioni, Tobias Marschall, Felix Mölder, Amir Niknejad, Lukasz Raczkowski, Marcel Reinders, Jeroen de Ridder, Antoine-Emmanuel Saliba, Antonios Somarakis, Oliver Stegle, Fabian J. Theis, Huan Yang, Alex Zelikovsky, Alice C. McHardy, Benjamin J. Raphael, Sohrab P. Shah, and Alexander Schönhuth (2020), "Eleven grand challenges in single-cell data science", Genome Biology 21: 31. Pubmed, doi: 10.1186/s13059-020-1926-6.
The recent boom in microfluidics and combinatorial indexing strategies, combined with low sequencing costs, has empowered single-cell sequencing technology. Thousands - or even millions - of cells analyzed in a single experiment amount to a data revolution in single-cell biology and pose unique data science problems. Here, we outline eleven challenges that will be central to bringing this emerging field of single-cell data science forward. For each challenge, we highlight motivating research questions, review prior work, and formulate open problems. This compendium is for established researchers, newcomers, and students alike, highlighting interesting and rewarding problems for the coming years.
Rilquer Mascarenhas Silva, Flavia M.R. Hirata, Eduardo Freitas Moreira, Amanda Campos, Miguel Loiola, Kaike Reis, Amaro Trindade Silva, Felipe Barbosa, Lucas Salles, Rafael Menezes, Rafael Veiga, Felipe Hernandes Coutinho, Bas E. Dutilh, Paulo R. Guimarães Jr, Ana Paula A. Assis, Anderson Ara Souza, José Garcia Vivas Miranda, Roberto F.S. Andrade, Bruno Vilela, Pedro Milet Meirelles (2020), "Integrating computational methods to investigate the macroecology of microbiomes", Frontiers in Genetics 10: 1344 Pubmed, doi: 10.3389/fgene.2019.01344.
Studies in microbiology have long been mostly restricted to small spatial scales. However, recent technological advances, such as new sequencing methodologies, have ushered an era of large-scale sequencing of environmental DNA data from multiple biomes worldwide. These global datasets can now be used to explore long standing questions of microbial ecology. New methodological approaches and concepts are being developed to study such large-scale patterns in microbial communities, resulting in new perspectives that represent a significant advances for both microbiology and macroecology. Here, we identify and review important conceptual, computational, and methodological challenges and opportunities in microbial macroecology. Specifically, we discuss the challenges of handling and analyzing large amounts of microbiome data to understand taxa distribution and co-occurrence patterns. We also discuss approaches for modeling microbial communities based on environmental data, including information on biological interactions to make full use of available Big Data. Finally, we summarize the methods presented in a general approach aimed to aid microbiologists in addressing fundamental questions in microbial macroecology, including classical propositions (such as "everything is everywhere, but the environment selects") as well as applied ecological problems, such as those posed by human induced global environmental changes.
Marleen Balvert, Georgios Patoulidis, Andrew Patti, Timo M. Deist, Christine Eyler, Bas E. Dutilh, Alexander Schönhuth, and David Craft (2020), "A Drug Recommendation System (Dr.S) for cancer cell lines", arXiv
Personalizing drug prescriptions in cancer care based on genomic information requires associating genomic markers with treatment effects. This is an unsolved challenge requiring genomic patient data in yet unavailable volumes as well as appropriate quantitative methods. We attempt to solve this challenge for an experimental proxy for which sufficient data is available: 42 drugs tested on 1018 cancer cell lines. Our goal is to develop a method to identify the drug that is most promising based on a cell line's genomic information. For this, we need to identify for each drug the machine learning method, choice of hyperparameters and genomic features for optimal predictive performance. We extensively compare combinations of gene sets (both curated and random), genetic features, and machine learning algorithms for all 42 drugs. For each drug, the best performing combination (considering only the curated gene sets) is selected. We use these top model parameters for each drug to build and demonstrate a Drug Recommendation System (Dr.S). Insights resulting from this analysis are formulated as best practices for developing drug recommendation systems. The complete software system, called the Cell Line Analyzer, is written in Python and available on github.
|
There are a billion times more viruses on earth than there are stars in the universe. The most numerous are the bacteriophages, viruses that infect bacteria. For as long as bacteria have existed, they have been embroiled in an evolutionary battle: bacteriophages infect bacteria and bacteria try to avoid infection. This global equilibrium is billions of years old and is perhaps the most complex system in existence. With ever more interest in bacteriophages in both fundamental and applied research, the key question is this: can we predict how the interaction between bacteriophages and bacteria will evolve under different conditions? With this ERC Consolidator grant, I will address this question by exploiting large DNA datasets from all over the world. |
|
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Stuart G. Siddell, Peter J. Walker, Elliot J. Lefkowitz, Arcady R. Mushegian, Bas E. Dutilh, Dutilh, Balázs Harrach, Robert L. Harrison, Sandra Junglen, Nick J. Knowles, Andrew M. Kropinski, Mart Krupovic, Jens H. Kuhn, Max L. Nibert, Luisa Rubino, Sead Sabanadzovic, Peter Simmonds, Arvind Varsani, Francisco Murilo Zerbini, and Andrew J. Davison (2019), "Binomial nomenclature for virus species: a consultation", Archives of Virology 165: 519-525, Pubmed, doi: 10.1007/s00705-019-04477-6. Erratum.
The Executive Committee of the International Committee on Taxonomy of Viruses (ICTV) recognizes the need for a standardized nomenclature for virus species. This article sets out the case for establishing a binomial nomenclature and presents the advantages and disadvantages of different naming formats. The Executive Committee understands that adopting a binomial system would have major practical consequences, and invites comments from the virology community before making any decisions to change the existing nomenclature. The Executive Committee will take account of these comments in deciding whether to approve a standardized binomial system at its next meeting in October 2020. Note that this system would relate only to the formal names of virus species and not to the names of viruses.
Patrick A. de Jonge, F.A. Bastiaan von Meijenfeldt, Laura E. van Rooijen, Stan J.J. Brouns and Bas E. Dutilh (2019) "Evolution of BACON domain tandem repeats in crAssphage and novel gut bacteriophage lineages", Viruses 11: 1085, doi: 10.3390/v11121085, Pubmed.
The human gut contains an expanse of largely unstudied bacteriophages. Among the most common are crAss-like phages, which were predicted to infect Bacteriodetes hosts. CrAssphage, the first crAss-like phage to be discovered, contains a protein encoding a Bacteroides-associated carbohydrate-binding often N-terminal (BACON) domain tandem repeat. Because protein domain tandem repeats are often hotspots of evolution, BACON domains may provide insight into the evolution of crAss-like phages. Here, we studied the biodiversity and evolution of BACON domains in bacteriophages by analysing over 2 million viral contigs. We found a high biodiversity of BACON in seven gut phage lineages, including five known crAss-like phage lineages and two novel gut phage lineages that are distantly related to crAss-like phages. In three BACON-containing phage lineages, we found that BACON domain tandem repeats were associated with phage tail proteins, suggestive of a possible role of these repeats in host binding. In contrast, individual BACON domains that did not occur in tandem were not found in the proximity of tail proteins. In two lineages, tail-associated BACON domain tandem repeats evolved largely through horizontal transfer of separate domains. In the third lineage that includes the prototypical crAssphage, the tandem repeats arose from several sequential domain duplications, resulting in a characteristic tandem array that is distinct from bacterial BACON domains. We conclude that phage tail-associated BACON domain tandem repeats have evolved in at least two independent cases in gut bacteriophages, including in the widespread gut phage crAssphage.
Parag D. Dabir, Carlijn E. Bruggeling, Rachel S. van der Post, Bas E. Dutilh, Nicoline Hoogerbrugge, Marjolijn J. L. Ligtenberg, Annemarie Boleij, and Iris D. Nagtegaal (2019) "Microsatellite instability screening in colorectal adenomas to detect Lynch syndrome patients? A systematic review and meta-analysis", European Journal of Human Genetics 28: 277-286, doi: 10.1038/s41431-019-0538-7, Pubmed.
The colorectal cancer spectrum has changed due to population screening programs, with a shift toward adenomas and early cancers. Whether it would be a feasible option to test these adenomas for detection of Lynch syndrome (LS) patients is unclear. Through meta-analysis and systematic review, risk factors for DNA mismatch repair deficiency (dMMR) and microsatellite instability (MSI) in adenomas were identified in LS and unselected patient cohorts. Data were extracted for patient age and MMR variant together with adenoma type, grade, size, and location. A total of 41 studies were included, and contained more than 519 LS patients and 1698 unselected patients with 1142 and 2213 adenomas respectively. dMMR/MSI was present in 69.5% of conventional adenomas in LS patients, compared with 2.8% in unselected patients. In the LS cohort, dMMR/MSI was more frequently present in patients older than 60 years (82% versus 54%). dMMR/MSI was also more common in villous adenomas (84%), adenomas over 1 cm (81%), and adenomas with high grade dysplasia (88%). No significant differences were observed for dMMR/MSI in relation to MMR variants and location of adenomas. In the context of screening, we conclude that detection of dMMR/MSI in conventional adenomas of unselected patients is uncommon and might be considered as indication for LS testing. Within the LS cohort, 69.5% of LS patients could have been detected through dMMR/MSI screening of their conventional adenomas.
F.A. Bastiaan von Meijenfeldt, Ksenia Arkhipova, Diego D. Cambuy, Felipe H. Coutinho, and Bas E. Dutilh (2019) "Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT", Genome Biology 20: 217, Pubmed, doi: 10.1186/s13059-019-1817-x.
Current-day metagenomics analyses increasingly involve de novo taxonomic classification of long DNA sequences and metagenome-assembled genomes. Here, we show that the conventional best-hit approach often leads to classifications that are too specific, especially when the sequences represent novel deep lineages. We present a classification method that integrates multiple signals to classify sequences (Contig Annotation Tool, CAT) and metagenome-assembled genomes (Bin Annotation Tool, BAT). Classifications are automatically made at low taxonomic ranks if closely related organisms are present in the reference database and at higher ranks otherwise. The result is a high classification precision even for sequences from considerably unknown organisms.
Paul Metz, Martijn J. Tjan, Shoaguang Wu, Mehrosh Pervaiz, Susanne Hermans, Aishwarya Shettigar, Cynthia L. Sears, Tina Ritschel, Bas E. Dutilh, and Annemarie Boleij (2019), "Drug discovery and repurposing inhibits a major gut pathogen-derived oncogenic toxin", Frontiers in Cellular and Infection Microbiology 9: 364, Pubmed, doi: 10.3389/fcimb.2019.00364.
Objective The human intestinal microbiome plays an important role in inflammatory bowel disease (IBD) and colorectal cancer (CRC) development. One of the first discovered bacterial mediators involves Bacteroides fragilis toxin (BFT, also named as fragilysin), a metalloprotease encoded by enterotoxigenic Bacteroides fragilis (ETBF) that causes barrier disruption and inflammation of the colon, leads to tumorigenesis in susceptible mice, and is enriched in the mucosa of IBD and CRC patients. Thus, targeted inhibition of BFT may benefit ETBF carrying patients. Design By applying two complementary in silico drug design techniques, drug repositioning and molecular docking, we predicted potential BFT inhibitory compounds. Top candidates were tested in vitro on the CRC epithelial cell line HT29/c1 for their potential to inhibit key aspects of BFT activity, being epithelial morphology changes, E-cadherin cleavage (a marker for barrier function) and increased IL-8 secretion. Results The primary bile acid and existing drug chenodeoxycholic acid (CDCA), currently used for treating gallstones, cerebrotendinous xanthomatosis, and constipation, was found to significantly inhibit all evaluated cell responses to BFT exposure. The inhibition of BFT resulted from a direct interaction between CDCA and BFT, as confirmed by an increase in the melting temperature of the BFT protein in the presence of CDCA. Conclusion Together, our results show the potential of in silico drug discovery to combat harmful human and microbiome-derived proteins and more specifically suggests a potential for retargeting CDCA to inhibit the pro-oncogenic toxin BFT.
Phages are increasingly recognized as important members of host-associated microbiomes, with a vast genomic diversity. The new frontier is to understand how phages may affect higher order processes, such as in the context of host-microbe interactions. Here, we use marine sponges as a model to investigate the interplay between phages, bacterial symbionts, and eukaryotic hosts. Using viral metagenomics, we find that sponges, although massively filtering seawater, harbor species-specific and even individually unique viral signatures that are taxonomically distinct from other environments. We further discover a symbiont phage-encoded ankyrin-domain-containing protein, which is widely spread in phages of many host-associated contexts including human. We confirm in macrophage infection assays that the ankyrin protein (ANKp) modulates the eukaryotic host immune response against bacteria. We predict that the role of ANKp in nature is to facilitate coexistence in the tripartite interplay between phages, symbionts, and sponges and possibly many other host-microbe associations. |

|
Simon Roux, Jelle Matthijnssens, and Bas E. Dutilh (2019), "Metagenomics in Virology", Reference Module in Life Sciences. doi: 10.1016/B978-0-12-809633-8.20957-6.
Metagenomics, i.e., the sequencing and analysis of genomic information extracted directly from clinical or environmental samples, has become a fundamental tool to explore the viral world. Against the background of an extensive viral diversity revealed by metagenomics across many environments, new sequence assembly approaches that reconstruct complete genome sequences from metagenomes have recently revealed surprisingly cosmopolitan viruses in specific ecological niches. Metagenomics is also applied to clinical samples as a non-targeted diagnostic and surveillance tool. By enabling the study of these uncultivated viruses, metagenomics provides invaluable insights into the virus-host interactions, epidemiology, ecology, and evolution of viruses across all ecosystems.
Peter J. Walker, Stuart G. Siddell, Elliot J. Lefkowitz, Arcady R. Mushegian, Donald M. Dempsey, Bas E. Dutilh, Balázs Harrach, Robert L. Harrison, R. Curtis Hendrickson, Sandra Junglen, Nick J. Knowles, Andrew M. Kropinski, Mart Krupovic, Jens H. Kuhn, Max Nibert, Luisa Rubino, Sead Sabanadzovic, Peter Simmonds, Arvind Varsani, Francisco Murilo Zerbini, and Andrew J. Davison (2019), "Changes to virus taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2019)", Archives of Virology 164: 2417-2429. Pubmed, doi: 10.1007/s00705-019-04306-w.
This article reports the changes to virus taxonomy approved and ratified by the International Committee on Taxonomy of Viruses (ICTV) in February 2019. Of note, in addition to seven new virus families, the ICTV has approved, by an absolute majority, the creation of the realm Riboviria, a likely monophyletic group encompassing all viruses with positive-strand, negative-strand and double-strand genomic RNA that use cognate RNA-directed RNA polymerases for replication.
Bojian Yin*, Marleen Balvert*, Rick A. A. van der Spek, Bas E. Dutilh, Sander Bohté, Jan Veldink, and Alexander Schönhuth (2019), "Using the structure of genome data in the design of deep neural networks for predicting amyotrophic lateral sclerosis from genotype", Bioinformatics 35: i538-i547. Pubmed, doi: 10.1093/bioinformatics/btz369. *Authors contributed equally.
Motivation Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease caused by aberrations in the genome. While several disease-causing variants have been identified, a major part of heritability remains unexplained. ALS is believed to have a complex genetic basis where non-additive combinations of variants constitute disease, which cannot be picked up using the linear models employed in classical genotype-phenotype association studies. Deep learning on the other hand is highly promising for identifying such complex relations. We therefore developed a deep-learning based approach for the classification of ALS patients versus healthy individuals from the Dutch cohort of the Project MinE dataset. Based on recent insight that regulatory regions harbor the majority of disease-associated variants, we employ a two-step approach: first promoter regions that are likely associated to ALS are identified, and second individuals are classified based on their genotype in the selected genomic regions. Both steps employ a deep convolutional neural network. The network architecture accounts for the structure of genome data by applying convolution only to parts of the data where this makes sense from a genomics perspective. Results Our approach identifies potentially ALS-associated promoter regions, and generally outperforms other classification methods. Test results support the hypothesis that non-additive combinations of variants contribute to ALS. Architectures and protocols developed are tailored toward processing population-scale, whole-genome data. We consider this a relevant first step toward deep learning assisted genotype-phenotype association in whole genome-sized data. Availability and implementation Our code will be available on Github, together with a synthetic dataset (https://github.com/byin-cwi/ALS-Deeplearning). The data used in this study is available to bona-fide researchers upon request.
Microbiomes are vast communities of microorganisms and viruses that populate all natural ecosystems. Viruses have been considered to be the most variable component of microbiomes, as supported by virome surveys and examples of high genomic mosaicism. However, recent evidence suggests that the human gut virome is remarkably stable compared with that of other environments. Here, we investigate the origin, evolution and epidemiology of crAssphage, a widespread human gut virus. Through a global collaboration, we obtained DNA sequences of crAssphage from more than one-third of the world's countries and showed that the phylogeography of crAssphage is locally clustered within countries, cities and individuals. We also found fully colinear crAssphage-like genomes in both Old-World and New-World primates, suggesting that the association of crAssphage with primates may be millions of years old. Finally, by exploiting a large cohort of more than 1,000 individuals, we tested whether crAssphage is associated with bacterial taxonomic groups of the gut microbiome, diverse human health parameters and a wide range of dietary factors. We identified strong correlations with different clades of bacteria that are related to Bacteroidetes and weak associations with several diet categories, but no significant association with health or disease. We conclude that crAssphage is a benign cosmopolitan virus that may have coevolved with the human lineage and is an integral part of the normal human gut virome. |

We found crAssphage in over one third of all the countries in the world. Illustration by Bas E. Dutilh. |
Jakub Barylski, François Enault, Bas E. Dutilh, Margo B.P. Schuller, Robert A. Edwards, Annika Gillis, Jochen Klumpp, Petar Knezevic, Mart Krupovic, Jens H. Kuhn, Rob Lavigne, Hanna M. Oksanen, Matthew B. Sullivan, Ho bin Jang, Peter Simmonds, Pakorn Aiewsakun, Johannes Wittmann, Igor Tolstoy, J. Rodney Brister, Andrew M. Kropinski, and Evelien M. Adriaenssens (2019), "Analysis of Spounaviruses as a case study for the overdue reclassification of tailed phages", Systematic Biology 69: 110-123, (preprint), Pubmed, doi: 10.1093/sysbio/syz036.
Tailed bacteriophages are the most abundant and diverse viruses in the world, with genome sizes ranging from 10 kbp to over 500 kbp.
Yet, due to historical reasons, all this diversity is confined to a single virus order - Caudovirales, composed of just four families: Myoviridae, Siphoviridae, Podoviridae, and the newly created Ackermannviridae family.
In recent years this morphology-based classification scheme has started to crumble under the constant flood of phage sequences, revealing that tailed phages are even more genetically diverse than once thought.
This prompted us, the Bacterial and Archaeal Viruses Subcommittee of the International Committee on Taxonomy of Viruses (ICTV) to consider overall reorganization of phage taxonomy.
In this study, we used a wide range of complementary methods - including comparative genomics, core genome analysis, and marker gene phylogenetics - to show that the group of Bacillus phage SPO1-related viruses previously classified into the Spounavirinae subfamily, is clearly distinct from other members of the family Myoviridae and its diversity deserves the rank of an autonomous family.
Thus, we removed this group from the Myoviridae family and created the family Herelleviridae - a new taxon of the same rank.
In the process of the taxon evaluation we explored the feasibility of different demarcation criteria and critically evaluated the usefulness of our methods for phage classification.
The convergence of results, drawing a consistent and comprehensive picture of a new family with associated subfamilies, regardless of method demonstrates that the tools applied here are particularly useful in phage taxonomy.
We are convinced that creation of this novel family is a crucial milestone towards much-needed re-classification in the Caudovirales order.
|
The Third Annual Meeting of the European Virus Bioinformatics Center (EVBC) took place in Glasgow, United Kingdom, 28-29 March 2019. Virus bioinformatics has become central to virology research, and advances in bioinformatics have led to improved approaches to investigate viral infections and outbreaks, being successfully used to detect, control, and treat infections of humans and animals. This active field of research has attracted approximately 110 experts in virology and bioinformatics/computational biology from Europe and other parts of the world to attend the two-day meeting in Glasgow to increase scientific exchange between laboratory- and computer-based researchers. The meeting was held at the McIntyre Building of the University of Glasgow; a perfect location, as it was originally built to be a place for "rubbing your brains with those of other people", as Rector Stanley Baldwin described it. The goal of the meeting was to provide a meaningful and interactive scientific environment to promote discussion and collaboration and to inspire and suggest new research directions and questions. The meeting featured eight invited and twelve contributed talks, on the four main topics: (1) systems virology, (2) virus-host interactions and the virome, (3) virus classification and evolution and (4) epidemiology, surveillance and evolution. Further, the meeting featured 34 oral poster presentations, all of which focused on specific areas of virus bioinformatics. This report summarizes the main research findings and highlights presented at the meeting. |

|
Guanabara Bay is a tropical estuarine ecosystem that receives massive anthropogenic impacts from the metropolitan region of Rio de Janeiro. This ecosystem suffers from an ongoing eutrophication process that has been shown to promote the emergence of potentially pathogenic bacteria, giving rise to public health concerns. Although previous studies have investigated how environmental parameters influence the microbial community of Guanabara Bay, they often have been limited to small spatial and temporal gradients and have not been integrated into predictive mathematical models. Our objective was to fill this knowledge gap by building models that could predict how temperature, salinity, phosphorus, nitrogen and transparency work together to regulate the abundance of bacteria, chlorophyll and Vibrio (a potential human pathogen) in Guanabara Bay. To that end, we built artificial neural networks to model the associations between these variables. These networks were carefully validated to ensure that they could provide accurate predictions without biases or overfitting. The estimated models displayed high predictive capacity (Pearson correlation coefficients >=0.67 and root mean square error <=0.55). Our findings showed that temperature and salinity were often the most important factors regulating the abundance of bacteria, chlorophyll and Vibrio (absolute importance >=5) and that each of these has a unique level of dependence on nitrogen and phosphorus for their growth. These models allowed us to estimate the Guanabara Bay microbiome's response to changes in environmental conditions, which allowed us to propose strategies for the management and remediation of Guanabara Bay. |

|
Microbes drive most ecosystems and are modulated by viruses that impact their lifespan, gene flow, and metabolic outputs. However, ecosystem-level impacts of viral community diversity remain difficult to assess due to classification issues and few reference genomes. Here, we establish an ~12-fold expanded global ocean DNA virome dataset of 195,728 viral populations, now including the Arctic Ocean, and validate that these populations form discrete genotypic clusters. Meta-community analyses revealed five ecological zones throughout the global ocean, including two distinct Arctic regions. Across the zones, local and global patterns and drivers in viral community diversity were established for both macrodiversity (inter-population diversity) and microdiversity (intra-population genetic variation). These patterns sometimes, but not always, paralleled those from macro-organisms and revealed temperate and tropical surface waters and the Arctic as biodiversity hotspots and mechanistic hypotheses to explain them. Such further understanding of ocean viruses is critical for broader inclusion in ecosystem models. |

|
Stuart G. Siddell, Peter J. Walker, Elliot J. Lefkowitz, Arcady R. Mushegian, Michael J. Adams, Bas E. Dutilh, Alexander E. Gorbalenya, Balázs Harrach, Robert L. Harrison, Sandra Junglen, Nick J. Knowles, Andrew M. Kropinski, Mart Krupovic, Jens H. Kuhn, Max Nibert, Luisa Rubino, Sead Sabanadzovic, Hélène Sanfaçon, Peter Simmonds, Arvind Varsani, Francisco Murilo Zerbini, and Andrew J. Davison (2018), "Additional changes to taxonomy ratified in a special vote by the International Committee on Taxonomy of Viruses (October 2018)", Archives of Virology 164: 943-946, doi: doi.org/10.1007/s00705-018-04136-2. Pubmed.
This article reports the changes to virus taxonomy approved and ratified by the International Committee on Taxonomy of Viruses (ICTV) in October 2018. Of note, the ICTV has approved, by an absolute majority, the creation of additional taxonomical ranks above those recognized previously. A total of 15 ranks (realm, subrealm, kingdom, subkingdom, phylum, subphylum, class, subclass, order, suborder, family, subfamily, genus, subgenus, and species) are now available to encompass the entire spectrum of virus diversity. Classification at ranks above genus is not obligatory but can be used by the authors of new taxonomic proposals when scientific justification is provided.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
|
Simon Roux, Evelien M. Adriaenssens, Bas E. Dutilh, Eugene V. Koonin, Andrew M. Kropinski, Mart Krupovic, Jens H. Kuhn, Rob Lavigne, J. Rodney Brister, Arvind Varsani, Clara Amid, Ramy K. Aziz, Seth R. Bordenstein, Peer Bork, Mya Breitbart, Guy R. Cochrane, Rebecca A. Daly, Christelle Desnues, Melissa B. Duhaime, Joanne B. Emerson, François Enault, Jed A. Fuhrman, Pascal Hingamp, Philip Hugenholtz, Bonnie L. Hurwitz, Natalia N. Ivanova, Jessica M. Labonté, Kyung-Bum Lee, Rex R. Malmstrom, Manuel Martinez-Garcia, Ilene Karsch Mizrachi, Hiroyuki Ogata, David Páez-Espino, Marie-Agnès Petit, Catherine Putonti, Thomas Rattei, Alejandro Reyes, Francisco Rodriguez-Valera, Karyna Rosario, Lynn Schriml, Frederik Schulz, Grieg F. Steward, Matthew B. Sullivan, Shinichi Sunagawa, Curtis A. Suttle, Ben Temperton, Susannah G. Tringe, Rebecca Vega Thurber, Nicole S. Webster, Katrine L. Whiteson, Steven W. Wilhelm, K. Eric Wommack, Tanja Woyke, Kelly C. Wrighton, Pelin Yilmaz, Takashi Yoshida, Mark J. Young, Natalya Yutin, Lisa Zeigler Allen, Nikos C. Kyrpides, and Emiley A. Eloe-Fadrosh (2018), "Minimum Information about an Uncultivated Virus Genome (MIUViG)", Nature Biotechnology 37: 29-37. Pubmed, doi: 10.1038/nbt.4306. News: UBC, JGI. We present an extension of the Minimum Information about any (x) Sequence (MIxS) standard for reporting sequences of uncultivated virus genomes. Minimum Information about an Uncultivated Virus Genome (MIUViG) standards were developed within the Genomic Standards Consortium framework and include virus origin, genome quality, genome annotation, taxonomic classification, biogeographic distribution and in silico host prediction. Community-wide adoption of MIUViG standards, which complement the Minimum Information about a Single Amplified Genome (MISAG) and Metagenome-Assembled Genome (MIMAG) standards for uncultivated bacteria and archaea, will improve the reporting of uncultivated virus genomes in public databases. In turn, this should enable more robust comparative studies and a systematic exploration of the global virosphere. |

Illustration by Leah Pantéa. |
Marleen Balvert, Alexander Schonhuth, and Bas Dutilh (2018), "Metagenomic read clustering based on overlap graphs", ICCABS, doi: 10.1109/ICCABS.2018.8541957.
When sequencing a metagenome one obtains a set of reads that originate from a variety of species. Clustering these reads by species is valuable for assembly as well as for metagenomic interpretation. Here we present an overlap graph-based read clustering approach.
Thais L. Brito, Amanda B. Campos, F.A. Bastiaan von Meijenfeldt, Julio P. Daniel, Gabriella B. Ribeiro, Genivaldo G.Z. Silva, Diego V. Wilke, Daniela T. de Moraes, Bas E. Dutilh, Pedro M. Meirelles, Amaro E. Trindade-Silva (2018) "The gill-associated microbiome is the main source of wood plant polysaccharide hydrolases and secondary metabolite gene clusters in the mangrove shipworm Neoteredo reynei", PLoS ONE 13: e0200437. Pubmed, doi: 10.1371/journal.pone.0200437.
Teredinidae are a family of highly adapted wood-feeding and wood-boring bivalves, commonly known as shipworms, whose evolution is linked to the acquisition of cellulolytic gammaproteobacterial symbionts harbored in bacteriocytes within the gills. In the present work we applied metagenomics to characterize microbiomes of the gills and digestive tract of Neoteredo reynei, a mangrove-adapted shipworm species found over a large range of the Brazilian coast. Comparative metagenomics grouped the gill symbiont community of different N. reynei specimens, indicating closely related bacterial types are shared. Similarly, the intestine and digestive gland communities were related, yet were more diverse than and showed no overlap with the gill community. Annotation of assembled metagenomic contigs revealed that the gill symbiotic community of N. reynei encodes a plethora of plant cell wall polysaccharides degrading glycoside hydrolase encoding genes, and Biosynthetic Gene Clusters (BGCs). In contrast, the digestive tract microbiomes seem to play little role in wood digestion and secondary metabolites biosynthesis. Metagenome binning recovered the nearly complete genome sequences of two symbiotic Teredinibacter strains from the gills, a representative of Teredinibacter turnerae "clade I" strain, and a yet to be cultivated Teredinibacter sp. type. These Teredinibacter genomes, as well as un-binned gill-derived gammaproteobacteria contigs, also include an endo-β-1,4-xylanase/acetylxylan esterase multi-catalytic carbohydrate-active enzyme, and a trans-acyltransferase polyketide synthase (trans-AT PKS) gene cluster with the gene cassette for generating β-branching on complex polyketides. Finally, we use multivariate analyses to show that the secondary metabolome from the genomes of Teredinibacter representatives, including genomes binned from N. reynei gills' metagenomes presented herein, stands out within the Cellvibrionaceae family by size, and enrichments for polyketide, nonribosomal peptide and hybrid BGCs. Results presented here add to the growing characterization of shipworm symbiotic microbiomes and indicate that the N. reynei gill gammaproteobacterial community is a prolific source of biotechnologically relevant enzymes for wood-digestion and bioactive compounds production.
Lidya Chaidir, Carolien Ruesen, Bas E. Dutilh, Ahmad Rizal Ganiem, Anggriani Andryani, Lika Apriani, Martijn A. Huynen, Rovina Ruslami, Philip C. Hill, Reinout van Crevel, and Bachti Alisjahbana (2018) "Use of whole genome sequencing to predict Mycobacterium tuberculosis drug resistance in Indonesia", Journal of Global Antimicrobial Resistance 16: 170-177. Pubmed doi: 10.1016/j.jgar.2018.08.018.
Background: Whole genome sequencing (WGS) is rarely used for drug-resistance testing of Mycobacterium tuberculosis in high-endemic settings. We present the first study from Indonesia, which has the second highest tuberculosis (TB) burden worldwide, with less than 50% of drug-resistant cases currently detected. Methods: We applied WGS in strains from 322 adult HIV-negative TB patients. Phenotypic DST was done for a portion of patients. Results: Fifty-one isolates (15.8%) harboured drug resistance mutations, including 42 among 322 patients (13.0%) with no prior TB treatment. Eight (2.5%) isolates were multidrug-resistant (MDR), one was extensively drug-resistant (XDR). Most mutations were found in katG (n=18), pncA (n=18), rpoB (n=10), fabG1 (n=9) and embB (n=9). The agreement of WGS-based resistance and phenotypic drug susceptibility testing (DST) to first-line drugs was high for isoniazid, rifampicin, and streptomycin, and less for ethambutol. Drug resistance was more common in Indo-Oceanic lineage strains (37.5%) than Euro-American (18.2%) and East-Asian lineage strains (10.3%; p=0.044), but combinations of multiple mutations were most common among East-Asian lineage strains (p=0.054). Conclusions: Our data support the potential use of WGS for more rapid and comprehensive prediction ofDR-TB in Indonesia. Future studies should address potential barriers in implementing WGS, the distribution of specific resistance mutations, and associations of particular mutations with endemic M. tuberculosis lineages in Indonesia.
Patrick A. de Jonge, Franklin L. Nobrega, Stan J.J. Brouns*, and Bas E. Dutilh* (2018) "Molecular and evolutionary determinants of bacteriophage host range", Trends in Microbiology 27: 51-63. Pubmed, doi: 10.1016/j.tim.2018.08.006. * Equal contributions.
The host range of a bacteriophage is the taxonomic diversity of hosts it can successfully infect. Host range, one of the central traits to understand in phages, is determined by a range of molecular interactions between phage and host throughout the infection cycle. While many well studied model phages seem to exhibit a narrow host range, recent ecological and metagenomics studies indicate that phages may have specificities that range from narrow to broad. There is a growing body of studies on the molecular mechanisms that enable phages to infect multiple hosts. These mechanisms, and their evolution, are of considerable importance to understanding phage ecology and the various clinical, industrial, and biotechnological applications of phage. Here we review knowledge of the molecular mechanisms that determine host range, provide a framework defining broad host range in an evolutionary context, and highlight areas for additional research.
Franklin L. Nobrega, Marnix Vlot, Patrick A. de Jonge, Lisa L. Dreesens, Hubertus J.E. Beaumont, Rob Lavigne, Bas E. Dutilh, and Stan J.J. Brouns (2018) "Targeting mechanisms of tailed bacteriophages", Nature Reviews Microbiology 16: 760-773. Pubmed, doi: 10.1038/s41579-018-0070-8.
Phages differ substantially in the bacterial hosts that they infect. Their host range is determined by the specific structures that they use to target bacterial cells. Tailed phages use a broad range of receptor-binding proteins, such as tail fibres, tail spikes and the central tail spike, to target their cognate bacterial cell surface receptors. Recent technical advances and new structure-function insights have begun to unravel the molecular mechanisms and temporal dynamics that govern these interactions. Here, we review the current understanding of the targeting machinery and mechanisms of tailed phages. These new insights and approaches pave the way for the application of phages in medicine and biotechnology and enable deeper understanding of their ecology and evolution.
Rineke H. Steenbergen, Martin Oti, Rob ter Horst, Wilson Tat, Chris Neufeldt, Alexandr Belovodskiy, Tiing Tiing Chua, Woo Jung Cho, Michael Joyce, Bas E. Dutilh, and D. Lorne Tyrrell (2018), "Establishing normal metabolism and differentiation in hepatocellular carcinoma cells by culturing in adult human serum", Scientific Reports 8: 11685. Pubmed, doi: 10.1038/s41598-018-29763-2.
Tissue culture medium routinely contains fetal bovine serum (FBS). Here we show that culturing human hepatoma cells in their native, adult serum (human serum, HS) results in the restoration of key morphological and metabolic features of normal liver cells. When moved to HS, these cells show differential transcription of 22-32% of the genes, stop proliferating, and assume a hepatocyte-like morphology. Metabolic analysis shows that the Warburg-like metabolic profile, typical for FBS-cultured cells, is replaced by a diverse metabolic profile consistent with in vivo hepatocytes, including the formation of large lipid and glycogen stores, increased glycogenesis, increased beta-oxidation and ketogenesis, and decreased glycolysis. Finally, organ-specific functions are restored, including xenobiotics degradation and secretion of bile, VLDL and albumin. Thus, organ-specific functions are not necessarily lost in cell cultures, but might be merely suppressed in FBS. The effect of serum is often overseen in cell culture and we provide a detailed study in the changes that occur and provide insight in some of the serum components that may play a role in the establishment of the differentiated phenotype.
Andrew M.Q. King, Elliot J. Lefkowitz, Arcady R. Mushegian, Michael J. Adams, Bas E. Dutilh, Alexander E. Gorbalenya, Balázs Harrach, Robert L. Harrison, Sandra Junglen, Nick J. Knowles, Andrew M. Kropinski, Mart Krupovic, Jens H. Kuhn, Max L. Nibert, Luisa Rubino, Sead Sabanadzovic, Hélène Sanfaçon, Stuart G. Siddell, Peter Simmonds, Arvind Varsani, Francisco Murilo Zerbini, and Andrew J. Davison (2018), "Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2018)", Archives of Virology 163: 2601-2631. Pubmed, PDF, doi: 10.1007/s00705-018-3847-1.
This article lists the changes to virus taxonomy approved and ratified by the International Committee on Taxonomy of Viruses in February 2018. A total of 451 species, 69 genera, 11 subfamilies, 9 families and one new order were added to the taxonomy. The current totals at each taxonomic level now stand at 9 orders, 131 families, 46 subfamilies, 803 genera and 4853 species. A change was made to the International Code of Virus Classification and Nomenclature to allow the use of the names of people in taxon names under appropriate circumstances. An updated Master Species List incorporating the approved changes was released in March 2018 (https://talk.ictvonline.org/taxonomy/).
Bashar Ibrahim‡, Ksenia Arkhipova‡, Arno C. Andeweg, Susana Posada-Céspedes, François Enault, Arthur Gruber, Eugene V. Koonin, Anne Kupczok, Philippe Lemey, Alice C. McHardy, Dino P. McMahon, Brett E. Pickett, David L. Robertson, Richard H. Scheuermann, Alexandra Zhernakova, Mark P. Zwart, Alexander Schönhuth‡, Bas E. Dutilh*‡, and Manja Marz*‡ (2018),
"Bioinformatics meets Virology: The European Virus Bioinformatics Center Second Annual Meeting",
Viruses 10: E256,
doi: 10.3390/v10050256.
PDF,
Pubmed.
*Contributed equally.
‡Organized the conference.
![]()
The Second Annual Meeting of the European Virus Bioinformatics Center (EVBC) held in Utrecht, The Netherlands focused on computational approaches in virology with topics including (but not limited to): virus discovery, diagnostics, (meta-)genomics, modeling, epidemiology, molecular structure, evolution, and viral ecology. The goals of the Second Annual Meeting were threefold: (i) to bring together virologists and bioinformaticians from across the academic, industrial, professional, and training sectors to share best practice; (ii) to provide a meaningful and interactive scientific environment to promote discussion and collaboration between students, postdoctoral fellows, and both new and established investigators; (iii) to inspire and suggest new research directions and questions. Approximately 120 researchers from around the world attended the Second Annual Meeting of the EVBC this year, including fifteen renowned international speakers. This report presents an overview of new developments and novel research findings that emerged during the meeting.
Daniel R. Garza, Marcel C. Van Verk, Martijn A. Huynen, and Bas E. Dutilh (2018), "Towards predicting the environmental metabolome from metagenomics with a mechanistic model", Nature Microbiology 3: 456-460, doi: 10.1038/s41564-018-0124-8. Pubmed. Behind the Paper. News: Cap Today, Radboudumc.
The environmental metabolome and metabolic potential of microbes are dominant and essential factors shaping microbial community composition. Recent advances genome annotation and systems biology now allow us to semi-automatically reconstruct genomescale metabolic models (GSMMs) of microbes based on their genome sequence. Next, growth of these models in a defined metabolic environment can be predicted in silico, mechanistically linking the metabolic fluxes of individual microbial populations to the community dynamics. A major advantage of GSMMs is that no training data is needed, besides information about the metabolic capacity of individual genes (genome annotation) and knowledge of the available environmental metabolites that allow the microbe to grow. However, the composition of the environment is often not fully determined and remains difficult to measure. We hypothesized that the relative abundance of different bacterial species, as measured by metagenomics, can be combined with GSMMs of individual bacteria to reveal the metabolic status of a given biome. Using a newly developed algorithm involving over 1,500 GSMMs of human-associated bacteria, we inferred distinct metabolomes for four human body-sites that are consistent with experimental data. Together, we link the metagenome to the metabolome in a mechanistic framework towards predictive microbiome modeling.
The Computational Pan-Genomics Consortium: Tobias Marschall, Manja Marz, Thomas Abeel, Louis Dijkstra, Bas E. Dutilh, Ali Ghaffaari, Paul Kersey, Wigard P. Kloosterman, Veli Mäkinen, Adam M. Novak, Benedict Paten, David Porubsky, Eric Rivals, Can Alkan, Jasmijn A. Baaijens, Paul I. W. De Bakker, Valentina Boeva, Raoul J. P. Bonnal, Francesca Chiaromonte, Rayan Chikhi, Francesca D. Ciccarelli, Robin Cijvat, Erwin Datema, Cornelia M. Van Duijn, Evan E. Eichler, Corinna Ernst, Eleazar Eskin, Erik Garrison, Mohammed El-Kebir, Gunnar W. Klau, Jan O. Korbel, Eric-Wubbo Lameijer, Benjamin Langmead, Marcel Martin, Paul Medvedev, John C. Mu, Pieter Neerincx, Klaasjan Ouwens, Pierre Peterlongo, Nadia Pisanti, Sven Rahmann, Ben Raphael, Knut Reinert, Dick de Ridder, Jeroen de Ridder, Matthias Schlesner, Ole Schulz-Trieglaff, Ashley D. Sanders, Siavash Sheikhizadeh, Carl Shneider, Sandra Smit, Daniel Valenzuela, Jiayin Wang, Lodewyk Wessels, Ying Zhang, Victor Guryev, Fabio Vandin, Kai Ye, and Alexander Schönhuth (2018), "Computational pan-genomics: status, promises and challenges", Briefings in Bioinformatics 19: 118-135, Pubmed, doi: 10.1093/bib/bbw089.
Many disciplines, from human genetics and oncology to plant breeding, microbiology and virology, commonly face the challenge of analyzing rapidly increasing numbers of genomes. In case of Homo sapiens, the number of sequenced genomes will approach hundreds of thousands in the next few years. Simply scaling up established bioinformatics pipelines will not be sufficient for leveraging the full potential of such rich genomic data sets. Instead, novel, qualitatively different computational methods and paradigms are needed. We will witness the rapid extension of computational pan-genomics, a new sub-area of research in computational biology. In this article, we generalize existing definitions and understand a pan-genome as any collection of genomic sequences to be analyzed jointly or to be used as a reference. We examine already available approaches to construct and use pan-genomes, discuss the potential benefits of future technologies and methodologies and review open challenges from the vantage point of the above-mentioned biological disciplines. As a prominent example for a computational paradigm shift, we particularly highlight the transition from the representation of reference genomes as strings to representations as graphs. We outline how this and other challenges from different application domains translate into common computational problems, point out relevant bioinformatics techniques and identify open problems in computer science. With this review, we aim to increase awareness that a joint approach to computational pan-genomics can help address many of the problems currently faced in various domains.
Carolien Ruesen, Lidya Chaidir, Arjan Van Laarhoven, Sofiati Dian, Ahmad Rizal Ganiem, Hanna Nebenzahl-Guimaraes, Martijn A. Huynen, Bachti Alisjahbana, Bas E. Dutilh, and Reinout van Crevel (2018) "Large-scale genomic analysis shows association between homoplastic genetic variation in Mycobacterium tuberculosis genes and meningeal or pulmonary tuberculosis", BMC Genomics 19: 122. Pubmed doi: 10.1186/s12864-018-4498-z.
Background: Meningitis is the most severe manifestation of tuberculosis. It is largely unknown why some people develop pulmonary TB (PTB) and others TB meningitis (TBM); we examined if the genetic background of infecting M. tuberculosis strains may be relevant. Methods: We whole-genome sequenced M. tuberculosis strains isolated from 322 HIV-negative tuberculosis patients from Indonesia and compared isolates from patients with TBM (n= 106) and PTB (n= 216). Using a phylogeny-adjusted genome-wide association method to count homoplasy events we examined phenotype-related changes at specific loci or genes in parallel branches of the phylogenetic tree. Enrichment scores for the TB phenotype were calculated on single nucleotide polymorphism (SNP), gene, and pathway level. Genetic associations were validated in an independent set of isolates. Results: Strains belonged to the East-Asian lineage (36.0%), Euro-American lineage (61.5%), and Indo-Oceaniclineage (2.5%). We found no association between lineage and phenotype (Chi-square=4.556; p=0.207). Largegenomic differences were observed between isolates; the minimum pairwise genetic distance varied from 17 to 689 SNPs. Using the phylogenetic tree, based on 28,544 common variable positions, we selected 54 TBM and 54 PTB isolates in terminal branch sets with distinct phenotypes. Genetic variation in Rv0218, and absence of Rv3343c, and nanK were significantly associated with disease phenotype in these terminal branch sets, and confirmed in the validation set of 214 unpaired isolates. Conclusions: Using homoplasy counting we identified genetic variation in three separate genes to be associated withthe TB phenotype, including one (Rv0218) which encodes a secreted protein that could play a role in host-pathogen interaction by altering pathogen recognition or acting as virulence effector.
Evelien M. Adriaenssens, Johannes Wittmann, Jens H. Kuhn, Dann Turner, Matthew B. Sullivan, Bas E. Dutilh, Ho Bin Jang, Leonardo J. van Zyl, Jochen Klumpp, Malgorzata Lobocka, Andrea I. Moreno Switt, Janis Rumnieks, Robert A. Edwards, Jumpei Uchiyama, Poliane Alfenas-Zerbini, Nicola K. Petty, Andrew M. Kropinski, Jakub Barylski, Annika Gillis, Martha R.C. Clokie, David Prangishvili, Rob Lavigne, Ramy K. Aziz, Siobain Duffy, Mart Krupovic, Minna M. Poranen, Petar Knezevic, Francois Enault, Yigang Tong, Hanna M. Oksanen, and J. Rodney Brister (2018), "Taxonomy of prokaryotic viruses: 2017 update from the ICTV Bacterial and Archaeal Viruses Subcommittee", Archives of Virology 163: 1125-1129. Pubmed, PDF, doi: 10.1007/s00705-018-3723-z.
Federica Gigliucci, F.A. Bastiaan von Meijenfeldt, Arnold Knijn, Valeria Michelacci, Gaia Scavia, Fabio Minelli, Bas E. Dutilh, Hamideh M. Ahmad, Gerwin C. Raangs, Alex W. Friedrich, John W. Rossen, and Stefano Morabito (2018) "Metagenomic characterization of the human intestinal microbiota in faecal samples from STEC-infected patients", Frontiers in Cellular and Infection Microbiology 8: 25. Pubmed, doi: 10.3389/fcimb.2018.00025.
The human intestinal microbiota is a homeostatic ecosystem with a remarkable impact on human health and the disruption of this equilibrium leads to an increased susceptibility to infection by numerous pathogens. In this study, we used shotgun metagenomic sequencing and two different bioinformatics approaches, based on mapping of the reads onto databases and on the reconstruction of putative draft genomes, to investigate possible changes in the composition of the intestinal microbiota in samples from patients with Shiga Toxin-producing E. coli (STEC) infection compared to healthy and healed controls, collected during an outbreak caused by a STEC O26:H11 infection. Both the bioinformatic procedures used, produced similar result with a good resolution of the taxonomic profiles of the specimens. The stool samples collected from the STEC infected patients showed a lower abundance of the members of Bifidobacteriales and Clostridiales orders in comparison to controls where those microorganisms predominated. These differences seemed to correlate with the STEC infection although a flexion in the relative abundance of the Bifidobacterium genus, part of the Bifidobacteriales order, was observed also in samples from Crohn's disease patients, displaying a STEC-unrelated dysbiosis. The metagenomics also allowed to identify in the STEC positive samples, all the virulence traits present in the genomes of the STEC O26 that caused the outbreak as assessed through isolation of the epidemic strain and whole genome sequencing. The results shown represent a first evidence of the changes occurring in the intestinal microbiota of children in the course of STEC infection and indicate that metagenomics may be a promising tool for the culture-independent clinical diagnosis of the infection.
Ksenia Arkhipova, Timofey Skvortsov, John P. Quinn, John W. McGrath, Christopher C.R. Allen, Bas E. Dutilh, Yvonne McElarney, and Leonid A. Kulakov (2018) "Temporal dynamics of uncultured viruses: a new dimension in viral diversity", ISME Journal 12: 199-211. Pubmed, doi: 10.1038/ismej.2017.157.
Recent work has vastly expanded the known viral genomic sequence space, but the seasonal dynamics of viral populations at the genome level remain unexplored. Here we followed the viral community in a freshwater lake for 1 year using genome-resolved viral metagenomics, combined with detailed analyses of the viral community structure, associated bacterial populations and environmental variables. We reconstructed 8950 complete and partial viral genomes, the majority of which were not persistent in the lake throughout the year, but instead continuously succeeded each other. Temporal analysis of 732 viral genus-level clusters demonstrated that one-fifth were undetectable at specific periods of the year. Based on host predictions for a subset of reconstructed viral genomes, we for the first time reveal three distinct patterns of host-pathogen dynamics, where the viruses may peak before, during or after the peak in their host's abundance, providing new possibilities for modelling of their interactions. Time series metagenomics opens up a new dimension in viral profiling, which is essential to understand the full scale of viral diversity and evolution, and the ecological roles of these important factors in the global ecosystem.
Katelyn McNair, Ramy K. Aziz, Gordon D. Pusch, Ross Overbeek, Bas E. Dutilh, and Robert Edwards (2018) "Phage genome annotation using the RAST pipeline", Methods in Molecular Biology 1681: 231-238. Pubmed, doi: 10.1007/978-1-4939-7343-9_17.
Phages are complex biomolecular machineries that have to survive in a bacterial world. Phage genomes show many adaptations to their lifestyle such as shorter genes, reduced capacity for redundant DNA sequences, and the inclusion of tRNAs in their genomes. In addition, phages are not free-living, they require a host for replication and survival. These unique adaptations provide challenges for the bioinformatics analysis of phage genomes. In particular, ORF calling, genome annotation, noncoding RNA (ncRNA) identification, and the identification of transposons and insertions are all complicated in phage genome analysis. We provide a road map through the phage genome annotation pipeline, and discuss the challenges and solutions for phage genome annotation as we have implemented in the rapid annotation using subsystems (RAST) pipeline.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Juline M. Walter, Felipe H. Coutinho, Bas E. Dutilh, Jean Swings, Fabiano Thompson, and Cristiane C. Thompson (2017), "Ecogenomics and taxonomy of Cyanobacteria phylum", Frontiers in Microbiology 8: 2132. Pubmed, doi: 10.3389/fmicb.2017.02132.
Cyanobacteria are major contributors to global biogeochemical cycles. The genetic diversity among Cyanobacteria enables them to thrive across many habitats, although only a few studies have analysed the association of phylogenomic clades to specific environmental niches. In this study, we adopted an ecogenomics strategy with the aim to delineate ecological niche preferences of Cyanobacteria and integrate them to the genomic taxonomy of these bacteria. First, an appropriate phylogenomic framework was established using a set of genomic taxonomy signatures (including a tree based on conserved gene sequences, genome-to-genome distance, and average amino acid identity) to analyse ninety-nine publicly available cyanobacterial genomes. Next, the relative abundances of these genomes were determined throughout diverse global marine and freshwater ecosystems, using metagenomic data sets. The whole-genome-based taxonomy of the ninety-nine genomes allowed us to identify 57 (of which 28 are new genera) and 87 (of which 32 are new species) different cyanobacterial genera and species, respectively. The ecogenomic analysis allowed the distinction of three major ecological groups of Cyanobacteria (named as i. Low Temperature; ii. Low Temperature Copiotroph; and iii. High Temperature Oligotroph) that were coherently linked to the genomic taxonomy. This work establishes a new taxonomic framework for Cyanobacteria in the light of genomic taxonomy and ecogenomic approaches.
|
Bas E. Dutilh, Alejandro Reyes, Richard J. Hall, and Katrine L. Whiteson (2017) "Editorial: Virus discovery by metagenomics: the (im)possibilities", Frontiers in Microbiology 8: 1710. Pubmed, doi: 10.3389/fmicb.2017.01710. This Frontiers in Virology Research Topic showcases how metagenomic and bioinformatic approaches have been combined to discover, classify and characterize novel viruses. Since the late 1800s, the discovery of new viruses was a gradual process. Viruses were described one by one using a suite of techniques such as (electron) microscopy and viral culture. Investigators were usually interested in a disease state within an organism, and expeditions in viral ecology were rare. The advent of metagenomics using high-throughput sequencing has revolutionized not only the rate of virus discovery, but also the nature of the discoveries. For example, the viral ecology and etiology of many human diseases are being characterized, non-pathogenic viral commensals are ubiquitous, and the description of environmental viromes is making progress. |

Frontiers prepared this nice E-book with all the articles in our Topic! |
Matheus C.F. Albuquerque, Yasmijn van Herwaarden, Guus A.M. Kortman, Bas E. Dutilh, Tanya Bisseling, and Annemarie Boleij (2017) "Preservation of bacterial DNA in 10-year-old guaiac FOBT cards and FIT tubes", Journal of Clinical Pathology 70: 994-996. Pubmed, doi: 10.1136/jclinpath-2017-204592.
With great interest we read the article of Taylor et al in the Journal of Clinical Pathology regarding the use of guaiac faecal occult blood test (gFOBT) cards for microbiome studies. gFOBT cards were found to be an easy to use option for stool collection and gained results comparable to fresh stool, even when cards were stored for up to 3 years at ambient temperature before DNA extraction. We would like to share our experience that even after 10 years of storage, gFOBT cards and faecal immunochemical test (FIT) tubes can be used to study the microbiome.
Felipe H. Coutinho, Cynthia B. Silveira, Gustavo B. Gregoracci, Cristiane C. Thompson, Robert A. Edwards, Corina P.D. Brussaard, Bas E. Dutilh*, and Fabiano L. Thompson* (2017) "Marine viruses discovered via metagenomics shed light on viral strategies throughout the oceans", Nature Communications 8: 15955. Pubmed, doi: 10.1038/ncomms15955. *Authors contributed equally. I recommend reading this cautionary note on inferring viral strategies from abundance correlations, and our reply.
| Marine viruses are key drivers of host diversity, population dynamics and biogeochemical cycling and contribute to the daily flux of billions of tons of organic matter. Despite recent advancements in metagenomics, much of their biodiversity remains uncharacterized. Here we report a data set of 27,346 marine virome contigs that includes 44 complete genomes. These outnumber all currently known phage genomes in marine habitats and include members of previously uncharacterized lineages. We designed a new method for host prediction based on co-occurrence associations that reveals these viruses infect dominant members of the marine microbiome such as Prochlorococcus and Pelagibacter. A negative association between host abundance and the virus-to-host ratio supports the recently proposed Piggyback-the-Winner model of reduced phage lysis at higher host densities. An analysis of the abundance patterns of viruses throughout the oceans revealed how marine viral communities adapt to various seasonal, temperature and photic regimes according to targeted hosts and the diversity of auxiliary metabolic genes. |

|
Douwe S. Maat*, Tristan Biggs*, Claire Evans, Judith D.L. van Bleijswijk, Nicole N. van der Wel, Bas E. Dutilh, and Corina P.D. Brussaard (2017) "Characterization and temperature dependence of arctic Micromonas polaris viruses", Viruses 9: 134. Pubmed, doi: 10.3390/v9060134. *Authors contributed equally.
Global climate change-induced warming of the Artic seas is predicted to shift the phytoplankton community towards dominance of smaller-sized species due to global warming. Yet, little is known about their viral mortality agents despite the ecological importance of viruses regulating phytoplankton host dynamics and diversity. Here we report the isolation and basic characterization of four prasinoviruses infectious to the common Arctic picophytoplankter Micromonas. We furthermore assessed how temperature influenced viral infectivity and production. Phylogenetic analysis indicated that the putative double-stranded DNA (dsDNA) Micromonas polaris viruses (MpoVs) are prasinoviruses (Phycodnaviridae) of approximately 120 nm in particle size. One MpoV showed intrinsic differences to the other three viruses, i.e., larger genome size (205 ± 2 vs. 191 ± 3 Kb), broader host range, and longer latent period (39 vs. 18 h). Temperature increase shortened the latent periods (up to 50%), increased the burst size (up to 40%), and affected viral infectivity. However, the variability in response to temperature was high for the different viruses and host strains assessed, likely affecting the Arctic picoeukaryote community structure both in the short term (seasonal cycles) and long term (global warming).
Amir Mohaghegh Motlagh, Ananda S. Bhattacharjee, Felipe Hernandes Coutinho, Bas E. Dutilh, Sherwood R. Casjens, and Ramesh K. Goel (2017) "Insights of phage-host interaction in hypersaline ecosystem through metagenomics analyses", Frontiers in Microbiology 8: 352. Pubmed, doi: 10.3389/fmicb.2017.00352.
Bacteriophages, as the most abundant biological entities on Earth, place significant predation pressure on their hosts. This pressure plays a critical role in the evolution, diversity, and abundance of bacteria. In addition, phages modulate the genetic diversity of prokaryotic communities through the transfer of auxiliary metabolic genes. Various studies have been conducted in diverse ecosystems to understand phage-host interactions and their effects on prokaryote metabolism and community composition. However, hypersaline environments remain among the least studied ecosystems and the interaction between the phages and prokaryotes in these habitats is poorly understood. This study begins to fill this knowledge gap by analyzing bacteriophage-host interactions in the Great Salt Lake, the largest prehistoric hypersaline lake in the Western Hemisphere. Our metagenomics analyses allowed us to comprehensively identify the bacterial and phage communities with Proteobacteria, Firmicutes, and Bacteroidetes as the most dominant bacterial species and Siphoviridae, Myoviridae, and Podoviridae as the most dominant viral families found in the metagenomic sequences. We also characterized interactions between the phage and prokaryotic communities of Great Salt Lake and determined how these interactions possibly influence the community diversity, structure, and biogeochemical cycles. In addition, presence of prophages and their interaction with the prokaryotic host was studied and showed the possibility of prophage induction and subsequent infection of prokaryotic community present in the Great Salt Lake environment under different environmental stress factors. We found that carbon cycle was the most susceptible nutrient cycling pathways to prophage induction in the presence of environmental stresses. This study gives an enhanced snapshot of phage and prokaryote abundance and diversity as well as their interactions in a hypersaline complex ecosystem, which can pave the way for further research studies.
The Course "Principles and Trends in Genomics and Computational Biology" is a first collaborative e-learning project involving the Oswaldo Cruz Foundation (Brazil) and the Institut Pasteur (France). The project was idealized by Fiocruz and Pasteur researchers Carolina Mizuno, Sara Cuadros, Fabiano Pais and Victor Pylro
Recent advances in science are leading to a revision and reorientation of methods, allowing old and current issues to be addressed in a new perspective.
Next-generation sequencing, metagenomics, metatranscriptomics and all other "omics" are permitting a comparative analysis of biological systems, generating a large quantity of data and findings.
Despite this progress, these technologies have developed faster than our ability to analyze this large amounts of data.
In order to overcome this problem the course will enable students to get a working knowledge of various facets of molecular and computational biology, including genome structure and organization, introduction to Linux, besides other most advanced topics, such as transcriptomics and proteomics.
The course will comprise four modules to be offered independently. Each module will contain several lessons. The length of each module will be of one week. The modules will consist of text, video classes and monitored activities.
In addition, a last module with state-of-the-art talks related to the course theme will be used to inspire students looking for new challenging projects in the area.
Daan R. Speth, Ilias Lagkouvardos, Yong Wang, Pei-Yuan Qian, Bas E. Dutilh, and Mike S. M. Jetten (2017) "Draft genome of Scalindua rubra, obtained from the interface above the Discovery Deep Brine in the Red Sea, sheds light on potential salt adaptation strategies in anammox bacteria", Microbial Ecology 74: 1-5. Pubmed, doi: 10.1007/s00248-017-0929-7.
Several recent studies have indicated that members of the phylum Planctomycetes are abundantly present at the brine-seawater interface (BSI) above multiple brine pools in the Red Sea. Planctomycetes include bacteria capable of anaerobic ammonium oxidation (anammox). Here, we investigated the possibility of anammox at BSI sites using metagenomic shotgun sequencing of DNA obtained from the BSI above the Discovery Deep brine pool. Analysis of sequencing reads matching the 16S rRNA and hzsA genes confirmed presence of anammox bacteria of the genus Scalindua. Phylogenetic analysis of the 16S rRNA gene indicated that this Scalindua sp. belongs to a distinct group, separate from the anammox bacteria in the seawater column, that contains mostly sequences retrieved from high-salt environments. Using coverage- and composition-based binning, we extracted and assembled the draft genome of the dominant anammox bacterium. Comparative genomic analysis indicated that this Scalindua species uses compatible solutes for osmoadaptation, in contrast to other marine anammox bacteria that likely use a salt-in strategy. We propose the name Candidatus Scalindua rubra for this novel species, alluding to its discovery in the Red Sea.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Evelien M. Adriaenssens, Mart Krupovic, Petar Knezevic, Hans-Wolfgang Ackermann, Jakub Barylski, J. Rodney Brister, Martha R. C. Clokie, Siobain Duffy, Bas E. Dutilh, Robert A. Edwards, Francois Enault, Ho Bin Jang, Jochen Klumpp, Andrew M. Kropinski, Rob Lavigne, Minna M. Poranen, David Prangishvili, Janis Rumnieks, Matthew B. Sullivan, Johannes Wittmann, Hanna M. Oksanen, Annika Gillis, Jens H. Kuhn (2016), "Taxonomy of prokaryotic viruses: 2016 update from the ICTV bacterial and archaeal viruses subcommittee", Archives of Virology 162: 1153-1157. Pubmed, PDF, doi: 10.1007/s00705-016-3173-4.
Lavinia Gambelli, Geert Cremers, Rob Mesman, Simon Guerrero, Bas E. Dutilh, Mike S. Jetten, Huub J. Op den Camp, and Laura van Niftrik (2016), "Ultrastructure and viral metagenome of bacteriophages from an anaerobic methane oxidizing Methylomirabilis bioreactor enrichment culture", Frontiers in Microbiology 7: 1740. Pubmed, doi: 10.3389/fmicb.2016.01740.
With its capacity for anaerobic methane oxidation and denitrification, the bacterium Methylomirabilis oxyfera plays an important role in natural ecosystems. Its unique physiology can be exploited for more sustainable wastewater treatment technologies. However, operational stability of full-scale bioreactors can experience setbacks due to, for example, bacteriophage blooms. By shaping microbial communities through mortality, horizontal gene transfer and metabolic reprogramming, bacteriophages are important players in most ecosystems. Here, we analysed an infected Methylomirabilis sp. bioreactor enrichment culture using (advanced) electron microscopy, viral metagenomics and bioinformatics. Electron micrographs revealed four different viral morphotypes, one of which was observed to infect Methylomirabilis cells. The infected cells contained densely packed ~55 nm icosahedral bacteriophage particles with a putative internal membrane. Various stages of virion assembly were observed. Moreover, during the bacteriophage replication, the host cytoplasmic membrane appeared extremely patchy, which suggests that the bacteriophages may use host bacterial lipids to build their own putative internal membrane. The viral metagenome contained 1.87 million base pairs of assembled viral sequences, from which five putative complete viral genomes were assembled and manually annotated. Using bioinformatics analyses, we could not identify which viral genome belonged to the Methylomirabilis- infecting bacteriophage, in part because the obtained viral genome sequences were novel and unique to this reactor system. Taken together these results show that new bacteriophages can be detected in anaerobic cultivation systems and that the effect of bacteriophages on the microbial community in these systems is a topic for further study.
Simon Roux, Jennifer R. Brum, Bas E. Dutilh, Shinichi Sunagawa, Melissa B. Duhaime, Alexander Loy, Bonnie T. Poulos, Natalie Solonenko, Elena Lara, Julie Poulain, Stéphane Pesant, Stefanie Kandels-Lewis, Céline Dimier, Marc Picheral, Sarah Searson, Corinne Cruaud, Adriana Alberti, Carlos M. Duarte, Josep M. Gasol, Dolors Vaqué, Peer Bork, Silvia G. Acinas, Patrick Wincker, and Matthew B. Sullivan for Tara Oceans Coordinators (2016), "Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses", Nature 537: 689-693. Pubmed, doi: 10.1038/nature19366.
Ocean microbes drive biogeochemical cycling on a global scale. However, this cycling is constrained by viruses that affect community composition, metabolic activity, and evolutionary trajectories. Owing to challenges with the sampling and cultivation of viruses, genome-level viral diversity remains poorly described and grossly understudied, with less than 1% of observed surface-ocean viruses known. Here we assemble complete genomes and large genomic fragments from both surface- and deep-ocean viruses sampled during the Tara Oceans and Malaspina research expeditions, and analyse the resulting 'global ocean virome' dataset to present a global map of abundant, double-stranded DNA viruses complete with genomic and ecological contexts. A total of 15,222 epipelagic and mesopelagic viral populations were identified, comprising 867 viral clusters (defined as approximately genus-level groups). This roughly triples the number of known ocean viral populations and doubles the number of candidate bacterial and archaeal virus genera, providing a near-complete sampling of epipelagic communities at both the population and viral-cluster level. We found that 38 of the 867 viral clusters were locally or globally abundant, together accounting for nearly half of the viral populations in any global ocean virome sample. While two-thirds of these clusters represent newly described viruses lacking any cultivated representative, most could be computationally linked to dominant, ecologically relevant microbial hosts. Moreover, we identified 243 viral-encoded auxiliary metabolic genes, of which only 95 were previously known. Deeper analyses of four of these auxiliary metabolic genes (dsrC, soxYZ, P-II (also known as glnB) and amoC) revealed that abundant viruses may directly manipulate sulfur and nitrogen cycling throughout the epipelagic ocean. This viral catalog and functional analyses provide a necessary foundation for the meaningful integration of viruses into ecosystem models where they act as key players in nutrient cycling and trophic networks.
News on Universiteit Utrecht (Nederlands/English), Ohio State University, University of Michigan, Universität Wien (Deutsch), Division of Microbial Ecology, Moore Foundation, Apa Science (Deutsch), BioPortfolio, Focus.it (Italiano), Krone.at (Deutsch), Kurier (Deutsch), Laboratory Equipment, Natural Science News, Nature World News, NRC (Nederlands), Phys.org, Science Daily, The Science Explorer, Science Orf (Deutsch), The Scientist, Seeker, Der Standard (Deutsch), Tendencias21 (Español), Vista al Mar (Español).
Felipe H. Coutinho, Bas E. Dutilh, Cristiane C. Thompson, and Fabiano L. Thompson (2016), "Proposal of fifteen new species of Parasynechococcus based on genomic, physiological and ecological features", Archives of Microbiology 198: 973-986. Pubmed, doi: 10.1007/s00203-016-1256-y.
Members of the recently proposed genus Parasynechococcus (Cyanobacteria) are extremely abundant throughout the global ocean and contribute significantly to global primary productivity. However, the taxonomy of these organisms remains poorly characterized. The aim of this study was to propose a new taxonomic framework for Parasynechococcus based on a genomic taxonomy approach that incorporates genomic, physiological and ecological data. Through in silico DNA-DNA hybridization, average amino acid identity, dinucleotide signatures and phylogenetic reconstruction, a total of 15 species of Parasynechococcus could be delineated. Each species was then described on the basis of their gene content, light and nutrient utilization strategies, geographical distribution patterns throughout the oceans and response to environmental parameters.
Adriana M. Fróes and Bas E. Dutilh (2016), "Bioinformatics for studying environmental microorganisms". In: Molecular Diversity of Environmental Prokaryotes. Eds. Thiago B. Rodrigues and Amaro E. Trindade Silva. CRC Press.
Daan R. Speth, Michiel H. in 't Zandt, Simon Guerrero-Cruz, Bas E. Dutilh*, and Mike S.M. Jetten* (2016), "Genome-based microbial ecology of anammox granules in a full-scale wastewater treatment system", Nature Communications 7: 11172. Pubmed, doi: 10.1038/ncomms11172. *Authors contributed equally. News on Bionieuws (Dutch), Water Online, Radboud University (English/Dutch).
Partial-nitritation anammox (PNA) is a novel wastewater treatment procedure for energy-efficient ammonium removal. Here we use genome-resolved metagenomics to build a genome-based ecological model of the microbial community in a full-scale PNA reactor. Sludge from the bioreactor examined here is used to seed reactors in wastewater treatment plants around the world; however, the role of most of its microbial community in ammonium removal remains unknown. Our analysis yielded 23 near-complete draft genomes that together represent the majority of the microbial community. We assign these genomes to distinct anaerobic and aerobic microbial communities. In the aerobic community, nitrifying organisms and heterotrophs predominate. In the anaerobic community, widespread potential for partial denitrification suggests a nitrite loop increases treatment efficiency. Of our genomes, 19 have no previously cultivated or sequenced close relatives and six belong to bacterial phyla without any cultivated members, including the most complete Omnitrophica (formerly OP3) genome to date.
Genivaldo Silva, Bas Dutilh, Robert Edwards (2016), "FOCUS2: agile and sensitive classification of metagenomics data using a reduced database", bioRxiv, doi: 10.1101/046425.
Summary: Metagenomics approaches rely on identifying the presence of organisms in the microbial community from a set of unknown DNA sequences. Sequence classification has valuable applications in multiple important areas of medical and environmental research. Here we introduce FOCUS2, an update of the previously published computational method FOCUS. FOCUS2 was tested with 10 simulated and 543 real metagenomes demonstrating that the program is more sensitive, faster, and more computationally efficient than existing methods. Availability: The Python implementation is freely available at https://edwards.sdsu.edu/FOCUS2.
Guus A.M. Kortman, Bas E. Dutilh, Annet J.H. Maathuis, Udo F. Engelke, Jos Boekhorst, Kevin P. Keegan, Fiona G.G. Nielsen, Jason Betley, Jacqueline C. Weir, Zoya Kingsbury, Leo A.J. Kluijtmans, Dorine W. Swinkels, Koen Venema, and Harold Tjalsma (2016), "Microbial metabolism shifts towards an adverse profile with supplementary iron in the TIM-2 in vitro model of the human colon", Frontiers in Microbiology 6: 1481. Pubmed, PDF, doi: 10.3389/fmicb.2015.01481.
Oral iron administration in African children can increase the risk for infections. However, it remains unclear to what extent supplementary iron affects the intestinal microbiome. We here explored the impact of iron preparations on microbial growth and metabolism in the well-controlled TNO's in vitro model of the large intestine (TIM-2). The model was inoculated with a human microbiota, without supplementary iron, or with 50 or 250 µmol/L ferrous sulfate, 50 or 250 µmol/L ferric citrate, or 50 µmol/L hemin. High resolution responses of the microbiota were examined by 16S rDNA pyrosequencing, microarray analysis, and metagenomic sequencing. The metabolome was assessed by fatty acid quantification, gas chromatography-mass spectrometry (GC-MS) and 1H-NMR spectroscopy. Cultured intestinal epithelial Caco-2 cells were used to assess fecal water toxicity. Microbiome analysis showed, among others, that supplementary iron induced decreased levels of Bifidobacteriaceae and Lactobacillaceae, while it caused higher levels of Roseburia and Prevotella. Metagenomic analyses showed an enrichment of microbial motility-chemotaxis systems, while the metabolome markedly changed from a saccharolytic to a proteolytic profile in response to iron. Branched chain fatty acids and ammonia levels increased significantly, in particular with ferrous sulfate. Importantly, the metabolite-containing effluent from iron-rich conditions showed increased cytotoxicity to Caco-2 cells. Our explorations indicate that in the absence of host influences, iron induces a more hostile environment characterized by a reduction of microbes that are generally beneficial, and increased levels of bacterial metabolites that can impair the barrier function of a cultured intestinal epithelial monolayer.
Mart Krupovic, Bas E. Dutilh, Evelien M. Adriaenssens, Johannes Wittmann, Finn K. Vogensen, Mathew B. Sullivan, Janis Rumnieks, David Prangishvili, Rob Lavigne, Andrew M. Kropinski, Jochen Klumpp, Annika Gillis, Francois Enault, Rob A. Edwards, Siobain Duffy, Martha R.C. Clokie, Jakub Barylski, Hans-Wolfgang Ackermann, and Jens H. Kuhn (2016), "Taxonomy of prokaryotic viruses: update from the ICTV bacterial and archaeal viruses subcommittee", Archives of Virology 161: 1095-1099. Pubmed, PDF, doi: 10.1007/s00705-015-2728-0. Prophage Blog.
|
Rob Lavigne, Takashi Yamada, Johannes Wittmann, Finn K. Vogensen, Mathew B. Sullivan, Janis Rumnieks, David Prangishvili, Jens H. Kuhn, Mart Krupovic, Andrew M. Kropinski, Jochen Klumpp, Annika Gillis, Francois Enault, Rob A. Edwards, Bas E. Dutilh, Siobain Duffy, Martha R.C. Clokie, Jakub Barylski, Hans-Wolfgang Ackermann, Evelien M. Adriaenssens (2015), "The Taxonomy of Bacterial & Archaeal Viruses: An Update from the International Committee on Taxonomy of Viruses", Evergreen Phage Meeting 2015, Evergreen, Washington, USA. |

|
Robert A. Edwards, Katelyn McNair, Karoline Faust, Jeroen Raes, and Bas E. Dutilh (2016), "Computational approaches to predict bacteriophage-host relationships", FEMS Microbiology Reviews fuv048, 40: 258-272. Pubmed, doi: 10.1093/femsre/fuv048. Editor's Choice.
Metagenomics has changed the face of virus discovery by enabling the accurate identification of viral genome sequences without requiring isolation of the viruses. As a result, metagenomic virus discovery leaves the first and most fundamental question about any novel virus unanswered: What host does the virus infect? The diversity of the global virosphere and the volumes of data obtained in metagenomic sequencing projects demand computational tools for virus-host prediction. We focus on bacteriophages (phages, viruses that infect bacteria), the most abundant and diverse group of viruses found in environmental metagenomes. By analyzing 820 phages with annotated hosts, we review and assess the predictive power of in silico phage-host signals. Sequence homology approaches are the most effective at identifying known phage-host pairs. Compositional and abundance-based methods contain significant signal for phage-host classification, providing opportunities for analyzing the unknowns in viral metagenomes. Together, these computational approaches further our knowledge of the interactions between phages and their hosts. Importantly, we find that all reviewed signals significantly link phages to their hosts, illustrating how current knowledge and insights about the interaction mechanisms and ecology of coevolving phages and bacteria can be exploited to predict phage-host relationships, with potential relevance for medical and industrial applications.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Daniela Bezemer, Anne Cori, Oliver Ratmann, Ard van Sighem, Hillegonda S. Hermanides, Bas E. Dutilh, Luuk Gras, Nuno Rodrigues Faria, Rob van den Hengel, Ashley J. Duits, Peter Reiss, Frank de Wolf, Christophe Fraser, and ATHENA observational cohort (2015), "Dispersion of the HIV-1 epidemic in men who have sex with men in The Netherlands: a combined mathematical model and phylogenetic analysis", PLoS Medicine 12: e1001898. Pubmed, PDF, doi: 10.1371/journal.pmed.1001898. News on NRC Medical News Today, Nederlands Dagblad, Seksoa, HIV Monitoring, Internazionale (Italiano), MedicalXpress, Science20.
Background The HIV-1 subtype B epidemic amongst men who have sex with men (MSM) is resurgent in many countries despite the widespread use of effective combination antiretroviral therapy (cART). In this combined mathematical and phylogenetic study of observational data, we aimed to find out the extent to which the resurgent epidemic is the result of newly introduced strains or of growth of already circulating strains. Methods and Findings As of November 2011, the ATHENA observational HIV cohort of all patients in care in the Netherlands since 1996 included HIV-1 subtype B polymerase sequences from 5,852 patients. Patients who were diagnosed between 1981 and 1995 were included in the cohort if they were still alive in 1996. The ten most similar sequences to each ATHENA sequence were selected from the Los Alamos HIV Sequence Database, and a phylogenetic tree was created of a total of 8,320 sequences. Large transmission clusters that included >=10 ATHENA sequences were selected, with a local support value >=0.9 and median pairwise patristic distance below the fifth percentile of distances in the whole tree. Time-varying reproduction numbers of the large MSM-majority clusters were estimated through mathematical modeling. We identified 106 large transmission clusters, including 3,061 (52%) ATHENA and 652 Los Alamos sequences. Half of the HIV sequences from MSM registered in the cohort in the Netherlands (2,128 of 4,288) were included in 91 large MSM-majority clusters. Strikingly, at least 54 (59%) of these 91 MSM-majority clusters were already circulating before 1996, when cART was introduced, and have persisted to the present. Overall, 1,226 (35%) of the 3,460 diagnoses among MSM since 1996 were found in these 54 long-standing clusters. The reproduction numbers of all large MSM-majority clusters were around the epidemic threshold value of one over the whole study period. A tendency towards higher numbers was visible in recent years, especially in the more recently introduced clusters. The mean age of MSM at diagnosis increased by 0.45 years/year within clusters, but new clusters appeared with lower mean age. Major strengths of this study are the high proportion of HIV-positive MSM with a sequence in this study and the combined application of phylogenetic and modeling approaches. Main limitations are the assumption that the sampled population is representative of the overall HIV-positive population and the assumption that the diagnosis interval distribution is similar between clusters. Conclusions The resurgent HIV epidemic amongst MSM in the Netherlands is driven by several large, persistent, self-sustaining, and, in many cases, growing sub-epidemics shifting towards new generations of MSM. Many of the sub-epidemics have been present since the early epidemic, to which new sub-epidemics are being added.
|
Daniela Bezemer, Oliver Ratmann, Ard van Sighem, Bas E. Dutilh, Nuno Faria, Rob van den Hengel, Luuk Gras, Peter Reiss, Frank de Wolf, Christophe Fraser, ATHENA observational cohort (2014), "Ongoing HIV-1 subtype B transmission networks in the Netherlands", CROI 2014, Boston, Massachusetts, USA. |
|
Genivaldo Gueiros Z. Silva, Kevin T. Green, Bas E. Dutilh, and Robert A. Edwards (2016), "SUPER-FOCUS: a tool for agile functional analysis of shotgun metagenomic data", Bioinformatics 32: 354-361. Pubmed, doi: 10.1093/bioinformatics/btv584.
Analyzing the functional profile of a microbial community from unannotated shotgun sequencing reads is one of the important goals in metagenomics. Functional profiling has valuable applications in biological research because it identifies the abundances of the functional genes of the organisms present in the original sample, answering the question what they can do. Currently available tools do not scale well with increasing data volumes, which is important because both the number and lengths of the reads produced by sequencing platforms keep increasing. Here we introduce SUPER-FOCUS, SUbsystems Profile by databasE Reduction using FOCUS, an agile homology-based approach using a reduced reference database to report the subsystems present in metagenomic datasets and profile their abundances. SUPER-FOCUS was tested with over 70 real metagenomes, the results showing that it accurately predicts the subsystems present in the profiled microbial communities, and is up to 1,000 times faster than other tools. Availability and implementation: SUPER-FOCUS was implemented in Python, and its source code and the tool website are freely available at https://edwards.sdsu.edu/SUPERFOCUS.
Qingzhen Hou, Bas E. Dutilh, Martijn A. Huynen, Jaap Heringa, and K. Anton Feenstra (2015), "Sequence specificity between interacting and non-interacting homologs identifies interface residues - a homodimer and monomer use case", BMC Bioinformatics 16: 325. Pubmed, PDF, doi: 10.1186/s12859-015-0758-y.
Background Protein families participating in protein-protein interactions may contain sub-families that have different binding characteristics, ranging from right binding to showing no interaction at all. Composition differences at the sequence level in these sub-families are often decisive to their differential functional interaction. Methods to predict interface sites from protein sequences typically exploit conservation as a signal. Here, instead, we provide proof of concept that the sequence specificity between interacting versus non-interacting groups can be exploited to recognise interaction sites. Results We collected homodimeric and monomeric proteins and formed homologous groups, each having an interacting (homodimer) subgroup and a non-interacting (monomer) subgroup. We then compiled multiple sequence alignments of the proteins in the homologous groups and identified compositional differences between the homodimeric and monomeric subgroups for each of the alignment positions. Our results show that this specificity signal distinguishes interface and other surface residues with 40.9 % recall and up to 25.1 % precision. Conclusions To our best knowledge, this is the first large scale study that exploits sequence specificity between interacting and non-interacting homologs to predict interaction sites from sequence information only. The performance obtained indicates that this signal contains valuable information to identify protein-protein interaction sites.
Jeremy J. Barr, Bas E. Dutilh, Connor T. Skennerton, Toshikazu Fukushima, Marcus L. Hastie, Jeffrey J. Gorman, Gene W. Tyson, and Philip L. Bond (2015), "Metagenomic and metaproteomic analyses of Accumulibacter phosphatis enriched floccular and granular biofilm", Environmental Microbiology 18: 273-287. Pubmed, doi: 10.1111/1462-2920.13019.
Biofilms are ubiquitous in nature, forming diverse adherent microbial communities that perform a plethora of functions. Here we operated two laboratory-scale sequencing batch reactors enriched with Candidatus Accumulibacter phosphatis (Accumulibacter) performing enhanced biological phosphorus removal (EBPR). Reactors formed two distinct biofilms, one floccular biofilm, consisting of small, loose, microbial aggregates, and one granular biofilm, forming larger, dense, spherical aggregates. Using metagenomic and metaproteomic methods we investigated the proteomic differences between these two biofilm communities, identifying a total of 2,022 unique proteins. To understand biofilm differences, we compared protein abundances that were statistically enriched in both biofilm states. Floccular biofilms were enriched with pathogenic secretion systems suggesting a highly competitive microbial community. Comparatively, granular biofilms revealed a high stress environment with evidence of nutrient starvation, phage predation pressure, and increased extracellular polymeric substance (EPS) and cell lysis. Granular biofilms were enriched in outer membrane transport proteins to scavenge the extracellular milieu for amino acids and other metabolites, likely released through cell lysis, to supplement metabolic pathways. This study provides the first detailed proteomic comparison between Accumulibacter-enriched floccular and granular biofilm communities, proposes a conceptual model for the granule biofilm, and offers novel insights into granule biofilm formation and stability.
Daniel R. Garza and Bas E. Dutilh (2015), "From cultured to uncultured genome sequences: metagenomics and modeling microbial ecosystems", Cellular and Molecular Life Sciences 72: 4287-4308. PDF, Pubmed, doi: 10.1007/s00018-015-2004-1.
Microorganisms and the viruses that infect them are the most numerous biological entities on Earth and enclose its greatest biodiversity and genetic reservoir. With strength in their numbers, these microscopic organisms are major players in the cycles of energy and matter that sustain all life. Scientists have only scratched the surface of this vast microbial world through culture-dependent methods. Recent developments in generating metagenomes, large random samples of nucleic acid sequences isolated directly from the environment, are providing comprehensive portraits of the composition, structure, and functioning of microbial communities. Moreover, advances in metagenomic analysis have created the possibility of obtaining complete or nearly complete genome sequences from uncultured microorganisms, providing important means to study their biology, ecology, and evolution. Here we review some of the recent developments in the field of metagenomics, focusing on the discovery of genetic novelty and on methods for obtaining uncultured genome sequences, including through the recycling of previously published datasets. Moreover we discuss how metagenomics has become a core scientific tool to characterize eco-evolutionary patterns of microbial ecosystems, thus allowing us to simultaneously discover new microbes and study their natural communities. We conclude by discussing general guidelines and challenges for modeling the interactions between uncultured microorganisms and viruses based on the information contained in their genome sequences. These models will significantly advance our understanding of the functioning of microbial ecosystems and the roles of microbes in the environment.
Felipe H. Coutinho, Pedro M. Meirelles, Ana Paula B. Moreira, Rodolfo P. Paranhos, Bas E. Dutilh, and Fabiano L. Thompson (2015), "Niche distribution and influence of environmental parameters in marine microbial communities: a systematic review", PeerJ 3: e1008. PDF, Pubmed.
Associations between microorganisms occur extensively throughout Earth's oceans. Understanding how microbial communities are assembled and how the presence or absence of species is related to that of others are central goals of microbial ecology. Here, we investigate co-occurrence associations between marine prokaryotes by combining 180 new and publicly available metagenomic datasets from different oceans in a large-scale meta-analysis. A co-occurrence network was created by calculating correlation scores between the abundances of microorganisms in metagenomes. A total of 1,906 correlations amongst 297 organisms were detected, segregating them into 11 major groups that occupy distinct ecological niches. Additionally, by analyzing the oceanographic parameters measured for a selected number of sampling sites, we characterized the influence of environmental variables over each of these 11 groups. Clustering organisms into groups of taxa that have similar ecology, allowed the detection of several significant correlations that could not be observed for the taxa individually.
Bas E. Dutilh (2015). "Illuminating 'dark matter': unknown bacteriophages and their role in our intestinal ecosystem", Vidi award, NWO.
This Vidi award from the Netherlands Organization for Scientific Research (NWO) enables me to do 5 years of independent research. I will discover new human gut-associated bacteriophages and investigate their role in structuring the gut microbiome.
T. David Matthews, Robert Schmieder, Genivaldo G. Z. Silva, Julia Busch, Noriko Cassman, Bas E. Dutilh, Dawn Green, Brian Matlock, Brian Heffernan, Gary J. Olsen, Leigh Farris Hanna, Dieter M, Schifferli, Stanley Maloy, Elizabeth A. Dinsdale, and Robert A. Edwards (2015), "Genomic comparison of the closely-related Salmonella enterica serovars Enteritidis, Dublin and Gallinarum", PLoS ONE 10: e0126883, PDF, Pubmed.
The Salmonella enterica serovars Enteritidis, Dublin, and Gallinarum are closely related but differ in virulence and host range. To identify the genetic elements responsible for these differences and to better understand how these serovars are evolving, we sequenced the genomes of Enteritidis strain LK5 and Dublin strain SARB12 and compared these genomes to the publicly available Enteritidis P125109, Dublin CT 02021853 and Dublin SD3246 genome sequences. We also compared the publicly available Gallinarum genome sequences from biotype Gallinarum 287/91 and Pullorum RKS5078. Using bioinformatic approaches, we identified single nucleotide polymorphisms, insertions, deletions, and differences in prophage and pseudogene content between strains belonging to the same serovar. Through our analysis we also identified several prophage cargo genes and pseudogenes that affect virulence and may contribute to a host-specific, systemic lifestyle. These results strongly argue that the Enteritidis, Dublin and Gallinarum serovars of Salmonella enterica evolve by acquiring new genes through horizontal gene transfer, followed by the formation of pseudogenes. The loss of genes necessary for a gastrointestinal lifestyle ultimately leads to a systemic lifestyle and niche exclusion in the host-specific serovars.
Julia Busch, Juliana R. Nascimento, Ana Carolina Magalhães, Bas E. Dutilh, and Elizabeth Dinsdale (2015), "Copper tolerance and distribution of epibiotic bacteria associated with giant kelp Macrocystis pyrifera in southern California", Ecotoxicology 24: 1131-1140, PDF, Pubmed.
Kelp forests in southern California are important ecosystems that provide habitat and nutrition to a multitude of species. Macrocystis pyrifera and other brown algae that dominate kelp forests, produce negatively charged polysaccharides on the cell surface, which have the ability to accumulate transition metals such as copper. Kelp forests near areas with high levels of boating and other industrial activities are exposed to increased amounts of these metals, leading to increased concentrations on the algal surface. The increased concentration of transition metals creates a harsh environment for colonizing microbes altering community structure. The impact of altered bacterial populations in the kelp forest have unknown consequences that could be harmful to the health of the ecosystem. In this study we describe the community of microorganisms associated with M. pyrifera, using a culture based approach, and their increasing tolerance to the transition metal, copper, across a gradient of human activity in southern California. The results support the hypothesis that M. pyrifera forms a distinct marine microhabitat and selects for species of bacteria that are rarer in the water column, and that copper-resistant isolates are selected for in locations with elevated exposure to transition metals associated with human activity.
Silke Appenzeller, Christian Gilissen, Jos Rijntjes, Bastiaan B.J. Tops, Annemiek Kastner-van Raaij, Konnie M. Hebeda, Loes Nissen, Bas E. Dutilh, J. Han J.M. van Krieken, and Patricia J.T.A. Groenen (2015), "Immunoglobulin rearrangement analysis from multiple lesions in the same patient using Next Generation Sequencing", Histopathology 67: 843-858. Pubmed, doi: 10.1111/his.12714.
Background For patients who have multiple lymphomas with discordant pathology, it is relevant to determine whether there is one disseminated lymphoma or two unrelated lymphomas. Patients with disseminated, clonally related lymphomas, are usually treated with the most powerful drugs available, while patients with unrelated (primary) lymphomas mostly receive standard first-line therapies. Methods We have used next generation sequencing on the Ion Torrent Personal Genome Machine to characterize the immunoglobulin heavy gene V-D-J rearrangements in two diagnostic tissue samples, including formalin-fixed and paraffin-embedded tissue, of two patients with iatrogenic immunodeficiency-associated Epstein-Barr virus lymphoproliferative disorder, with ulcerative colitis as underlying disease. Results The immunoglobulin rearrangement sequences obtained by next generation sequencing revealed undoubtedly clonally related lesions in two tissue biopsies that were taken over time in the first patient, which is concordant with disseminated lymphoma. The other patient showed two clonally unrelated lesions, which is incompatible with clonal dissemination. This information was not inferred from evaluation of the heavy and light chain rearrangements by fragment analysis, which is currently the gold standard. Conclusion Our study demonstrates the diagnostic application of next generation sequencing of immunoglobulin rearrangement assessment in pathology for clinical decision making in patients with several simultaneous or subsequent lymphoproliferations.
Richard J. Hall, Jenny L. Draper, Fiona G.G. Nielsen, and Bas E. Dutilh (2015), "Beyond research: a primer for considerations on using viral metagenomics in the field and clinic", Frontiers in Microbiology 6: 224, doi: 10.3389/fmicb.2015.00224. Pubmed, PDF. News on DNA Digest, UBC.
Powered by recent advances in next-generation sequencing technologies, metagenomics has already unveiled vast microbial biodiversity in a range of environments, and is increasingly being applied in clinics for difficult-to-diagnose cases. It can be tempting to suggest that metagenomics could be used as a "universal test" for all pathogens without the need to conduct lengthy serial testing using specific assays. While this is an exciting prospect, there are issues that need to be addressed before metagenomic methods can be applied with rigour as a diagnostic tool, including the potential for incidental findings, unforeseen consequences for trade and regulatory authorities, privacy and cultural issues, data sharing, and appropriate reporting of results to end-users. These issues will require consideration and discussion across a range of disciplines, including scientists, ethicists, clinicians, diagnosticians, health practitioners, and ultimately the public. Here, we provide a primer for consideration on some of these issues.
Daan R. Speth, Lina Russ, Boran Kartal, Huub J.M. Op den Camp, Bas E. Dutilh, and Mike S.M. Jetten (2015), "Draft genome sequence of anammox bacterium "Candidatus Scalindua brodae", obtained using differential coverage binning of sequencing data from two reactor enrichments", Genome Announcements 3: e01415-14, doi: 10.1128/genomeA.01415-14. Pubmed, PDF.
We present the draft genome of anammox bacterium "Candidatus Scalindua brodae", which at 282 contigs is a major improvement over the highly fragmented genome assembly of related species "Ca. Scalindua profunda" (1,580 contigs) which was previously published.
Cristiane C. Thompson, Gilda R. Amaral, Robert A. Edwards, Martin F. Polz, Bas E. Dutilh, David W. Ussery, Erko Stackebrandt, Jean Swings, and Fabiano L. Thompson (2015), "Microbial taxonomy in the post-genomic era: Rebuilding from scratch?", Archives of Microbiology 197: 359-370, doi: 10.1007/s00203-014-1071-2. Pubmed, PDF.
Prokaryotic taxonomy should provide adequate descriptions of prokaryotic diversity in ecological, clinical and industrial environments. Its cornerstone, the prokaryote species has been re-evaluated twice (Stackebrandt et al., 2002; Gevers et al., 2005). It is time to revisit polyphasic taxonomy (Vandamme & Peeters, 2014), its principles and its practice, including its underlying pragmatic species concept. Ultimately, we will be able to realize the old dream of our predecessors and build a genomic prokaryotic taxonomy with genome sequences as gold standards.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Bas E. Dutilh (2014), "Metagenomic ventures into outer sequence space", Bacteriophage 4: e979664. DOI, PDF.
Sequencing DNA or RNA directly from the environment often results in many sequencing reads that have no homologs in the database. These are referred to as "unknowns", and reflect the vast unexplored microbial sequence space of our biosphere, also known as "biological dark matter". However, unknowns also exist because metagenomic datasets are not optimally mined. There is a pressure on researchers to publish and move on, and the unknown sequences are often left for what they are, and conclusions drawn based on reads with annotated homologs. This can cause abundant and widespread genomes to be overlooked, such as the recently discovered human gut bacteriophage crAssphage. The unknowns may be enriched for bacteriophage sequences, the most abundant and genetically diverse component of the biosphere and of sequence space. However, it remains an open question, what is the actual size of biological sequence space? The de novo assembly of shotgun metagenomes is the most powerful tool to address this question.
Bas E. Dutilh, Noriko Cassman, Katelyn McNair, Savannah E. Sanchez, Genivaldo G.Z. Silva, Lance Boling, Jeremy J. Barr, Daan R. Speth, Victor Seguritan, Ramy K. Aziz, Ben Felts, Elizabeth A. Dinsdale, John L. Mokili and Robert A. Edwards (2014), "A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes", Nature Communications 5: 4498. Pubmed. Ranked in the 99 percentile of all tracked articles based on the Altmetric score.
Metagenomics, or sequencing of the genetic material from a complete microbial community, is a promising tool to discover novel microbes and viruses. Viral metagenomes typically contain many unknown sequences. Here we describe the discovery of a previously unidentified bacteriophage present in the majority of published human faecal metagenomes, which we refer to as crAssphage. Its ~97 kbp genome is six times more abundant in publicly available metagenomes than all other known phages together; it comprises up to 90% and 22% of all reads in virus-like particle (VLP)-derived metagenomes and total community metagenomes, respectively; and it totals 1.68% of all human faecal metagenomic sequencing reads in the public databases. The majority of crAssphage-encoded proteins match no known sequences in the database, which is why it was not detected before. Using a new co-occurrence profiling approach, we predict a Bacteroides host for this phage, consistent with Bacteroides-related protein homologues and a unique carbohydrate-binding domain encoded in the phage genome.
News about crAssphage: National Geographic Not Exactly Rocket Science, NPR Goats and Soda, Forbes, Science Friday (radio interview), BBC, BBC Mundo (Español), Nature Research Highlights. New Scientist 1, New Scientist 2, Scientific American, EOS (Nederlands), Twitter (#), Huffington Post, IFL Science!, NRC (Nederlands), nu.nl (Nederlands), Beta Sandwich (podcast minute 36), Austrian Tribune 1, Austrian Tribune 2, El Balad (Arabic), BayouBuzz, Bild (Deutsch), Bio-Medicine, BioPharma, Business Insider, Celebrity Cafe, China Topix, CN Blogs (Chinese), Cosmos, Cotidianul (Român), Counsel & Heal, Le Daily (Français), Daily Digest News, Daily Mail, Design & Trend, Diabetes.co.uk, Diario Panorama, 43.2 The Drop, DZ Online, East County Magazine, EBioTrade, Edwards lab, Einheit11, EurekAlert, Financial Express, Fox News, Franzcalvo, Frequency, GB Times, Gezondheid.nl (Nederlands), GGSOKU (Japanese), Il Giornale di Montesilvano (Italiano), Gizmodo, Google News, Google Trending terms, Guardian Liberty Voice, Health Canal, Health Hub, Healthline, Health Site, HNGN, HRN (Español), IANS Live, International Business Times 1, International Business Times 2, io9, Klaipėda (Lietuvos), KNAU, Knibmus, Kopalnia Wiedzy (Polski), KPCC, Kyle's Daily Bulletin, Lacto Bacto, Libertad Digital (Español), Live Science, Lockerdome, A man with a PhD, MaxiSciences (Français), Mejor Vendedor, Mendoza online (Español), Medical Daily, MedicalFacts.nl (Nederlands), MedicalXpress, medONLINE.at (Deutsch), Medportal.ru (русский), Meteoweb (Italiano), Microbiome Digest, Mizo News, Le Monde (Français), Mother Nature Network, MSFN, Muzic4you, Muy Interesante (Español), Nationale Zorggids (Nederlands), Nature Middle East, Nature World News, Newhub, News.de (Deutsch), NewsFactor, News Locker, News Tonight Africa, NineMSN, Noticias de la Ciencia (Español), La Nouvelle Tribune (Français), OnMed.gr (ελληνικα), Outbreak News Today, Pakistan Today, Le Point (Français), Prescripteurs (Français), Prevention, Radboudumc, R&D Magazine, Readable, Rectofossal Ambiguity, Reddit (AMA), Red Orbit, Regator, RIMLS, Salon, Science 2.0, Science Alert, Science Codex, Science Daily, Science Newsline, Science Sifter, Science World Report 1, Science World Report 2, The Scientist, Sci-News, Scinexx, SDSU, Slashdot, A Smaller Flea, Softpedia, Sohu (Chinese), Der Spiegel (Deutsch), Spire Healthcare Der Standard (Deutsch), TECHsme.sk (Slovenská), Tech Times, Tendencias (Español), Tendencias 21 (Español), The Times of India, Times of San Diego, Tinyletter, το χϖνι (ελληνικα), UBC, UK Wired, Vesti (русский), Virus Doctors blog, Voxxi, 90.9 wbur, Die Welt, WGBH News, Yahoo! News, Z News; and a Wikipedia page.
Genivaldo G.Z. Silva, Bas E. Dutilh, and Robert A. Edwards (2014), "FORMAL: A model to identify organisms present in metagenomes using Monte Carlo simulation". bioRXiv, doi: 10.1101/010801.
One of the major goals in metagenomics is to identify organisms present in the microbial community from a huge set of unknown DNA sequences. This profiling has valuable applications in multiple important areas of medical research such as disease diagnostics. Nevertheless, it is not a simple task, and many approaches that have been developed are slow and depend on the read length of the DNA sequences. Here we introduce an innovative and agile approach which k-mer and Monte Carlo simulation to profile and report abundant organisms present in metagenomic samples and their relative abundance without sequence length dependencies. The program was tested with a simulated metagenomes, and the results show that our approach predicts the organisms in microbial communities and their relative abundance.
Nelson Alves Junior, Pedro Milet Meirelles, Eidy de Oliveira Santos, Bas Dutilh, Genivaldo G.Z. Silva, Rodolfo Paranhos, Anderson S. Cabral, Carlos Rezende, Tetsuya Iida, Rodrigo L. de Moura, Ricardo Henrique Kruger, Renato C. Pereira, Rogério Valle, Tomoo Sawabe, Cristiane Thompson, and Fabiano Thompson (2014), "Microbial community diversity and physical-chemical features of the Southwestern Atlantic Ocean", Archives of Microbiology 197: 165-179, doi: 10.1007/s00203-014-1035-6. Pubmed, PDF.
Microbial oceanography studies have demonstrated the central role of microbes in functioning and nutrient cycling of the global ocean. Most of these former studies including at Southwestern Atlantic Ocean (SAO) focused on surface seawater and benthic organisms (e.g., coral reefs and sponges). This is the first metagenomic study of the SAO. The SAO harbors a great microbial diversity and marine life (e.g., coral reefs and rhodolith beds). The aim of this study was to characterize the microbial community diversity of the SAO along the depth continuum and different water masses by means of metagenomic, physical-chemical and biological analyses. The microbial community abundance and diversity appear to be strongly influenced by the temperature, dissolved organic carbon, and depth, and three groups were defined [1. surface waters; 2. sub-superficial chlorophyll maximum (SCM) (48-82 m) and 3. deep waters (236-1,200 m)] according to the microbial composition. The microbial communities of deep water masses [South Atlantic Central water, Antarctic Intermediate water and Upper Circumpolar Deep water] are highly similar. Of the 421,418 predicted genes for SAO metagenomes, 36.7 % had no homologous hits against 17,451,486 sequences from the North Atlantic, South Atlantic, North Pacific, South Pacific and Indian Oceans. From these unique genes from the SAO, only 6.64 % had hits against the NCBI non-redundant protein database. SAO microbial communities share genes with the global ocean in at least 70 cellular functions; however, more than a third of predicted SAO genes represent a unique gene pool in global ocean. This study was the first attempt to characterize the taxonomic and functional community diversity of different water masses at SAO and compare it with the microbial community diversity of the global ocean, and SAO had a significant portion of endemic gene diversity. Microbial communities of deep water masses (236-1,200 m) are highly similar, suggesting that these water masses have very similar microbiological attributes, despite the common knowledge that water masses determine prokaryotic community and are barriers to microbial dispersal. The present study also shows that SCM is a clearly differentiated layer within Tropical waters with higher abundance of phototrophic microbes and microbial diversity.
Bas E. Dutilh, Cristiane C. Thompson, Ana C.P. Vicente, Michel A. Marin, Clarence Lee, Genivaldo G.Z. Silva, Robert Schmieder, Bruno G.N. Andrade, Luciane Chimetto, Daniel Cuevas, Daniel Garza, Iruka N. Okeke, A. Oladipo Aboderin, Jessica Spangler, Tristen Ross, Elizabeth A. Dinsdale, Fabiano L. Thompson, Timothy T. Harkins and Robert A. Edwards (2014), "Comparative genomics of 274 Vibrio cholerae genomes reveals mobile functions structuring three niche dimensions", BMC Genomics 15: 654. Pubmed, PDF. Featured in Biome.
Background Vibrio cholerae is a globally dispersed pathogen that has evolved with humans for centuries, but also includes non-pathogenic environmental strains. Here, we identify the genomic variability underlying this remarkable persistence across the three major niche dimensions space, time, and habitat. Results Taking an innovative approach of genome-wide association applicable to microbial genomes (GWAS-M), we classify 274 complete V. cholerae genomes by niche, including 39 newly sequenced for this study with the Ion Torrent DNA-sequencing platform. Niche metadata were collected for each strain and analyzed together with comprehensive annotations of genetic and genomic attributes, including point mutations (single-nucleotide polymorphisms, SNPs), protein families, functions and prophages. Conclusions Our analysis revealed that genomic variations, in particular mobile functions including phages, prophages, transposable elements, and plasmids underlie the metadata structuring in each of the three niche dimensions. This underscores the role of phages and mobile elements as the most rapidly evolving elements in bacterial genomes, creating local endemicity (space), leading to temporal divergence (time), and allowing the invasion of new habitats. Together, we take a data-driven approach for comparative functional genomics that exploits high-volume genome sequencing and annotation, in conjunction with novel statistical and machine learning analyses to identify connections between genotype and phenotype on a genome-wide scale.
Bas E. Dutilh (2012), "Gene repertoires responsible for persistence of Vibrio cholerae across niche dimensions by Ion Torrent sequencing", talk at Advances in Genome Biology and Technology Conference (AGBT 2012), Marco Island, Florida, USA.
Yan Wei Lim, Daniel Cuevas, Genivaldo G.Z. Silva, Kristen Aguinaldo, Elizabeth Dinsdale, Andreas Haas, Mark Hatay, Savannah Sanchez, Linda Wegley-Kelly, Bas E. Dutilh, Timothy Harkins, Clarence Lee, Warren Tom, Stuart Sandin, Jennifer E. Smith, Brian Zgliczynski, Mark J.A. Vermeij, Forest Rohwer and Robert A. Edwards (2014), "Sequencing at sea: challenges and experiences in Ion Torrent PGM sequencing during the 2013 Southern Line Islands research expedition", PeerJ 2: e520. PDF. Selected for PeerJ Picks 2015.
Genomics and metagenomics have revolutionized our understanding of marine microbial ecology and the importance of microbes in global geochemical cycles. However, the process of DNA sequencing has always been an abstract extension of the research expedition, completed once the samples were returned to the laboratory. During the 2013 Southern Line Islands Research Expedition, we started the first effort to bring next generation sequencing to some of the most remote locations on our planet. We successfully sequenced twenty six marine microbial genomes,and two marine microbial metagenomes using the Ion Torrent PGM platform on the Merchant Yacht Hanse Explorer. Onboard sequence assembly, annotation, and analysis enabled us to investigate the role of the microbes in the coral reef ecology of these islands and atolls. This analysis identified phospohonate as an important phosphorous source for microbes growing in theLine Islands and reinforced the importance of L-serine in marine microbial ecosystems. Sequencing in the field allowed us to propose hypotheses and conduct experiments and further sampling based on the sequences generated. By eliminating the delay between sampling and sequencing, we enhanced the productivity of the research expedition. By overcoming the hurdles associated with sequencing on a boat in the middle of the Pacific Ocean we proved the flexibility of the sequencing, annotation, and analysis pipelines.
News highlights: EurekAlert, GenomeWeb, Genetic Engineering and Biotechnology News, International Business Times, Nature World News, Ocean Portal, PeerJ Blog, PeerJ Video, Science Daily, Tech Times, Times of San Diego.
Sarah Ghamrawi, Gilles Rénier, Patrick Saulnier, Stéphane Cuenot, Agata Zykwinska, Bas E. Dutilh, Christopher Thornton, Sébastien Faure and Jean-Philippe Bouchara (2014), "Cell wall modifications during conidial maturation of the human pathogenic fungus Pseudallescheria boydii". PLoS ONE 9: e100290. Pubmed, PDF.
Progress in extending the life expectancy of cystic fibrosis (CF) patients remains jeopardized by the increasing incidence of fungal respiratory infections. Pseudallescheria boydii (P. boydii), an emerging pathogen of humans, is a filamentous fungus frequently isolated from the respiratory secretions of CF patients. It is commonly believed that infection by this fungus occurs through inhalation of airborne conidia, but the mechanisms allowing the adherence of Pseudallescheria to the host epithelial cells and its escape from the host immune defenses remain largely unknown. Given that the cell wall orchestrates all these processes, we were interested in studying its dynamic changes in conidia as function of the age of cultures. We found that the surface hydrophobicity and electronegative charge of conidia increased with the age of culture. Melanin that can influence the cell surface properties, was extracted from conidia and estimated using UV-visible spectrophotometry. Cells were also directly examined and compared using electron paramagnetic resonance (EPR) that determines the production of free radicals. Consistent with the increased amount of melanin, the EPR signal intensity decreased suggesting polymerization of melanin. These results were confirmed by flow cytometry after studying the effect of melanin polymerization on the surface accessibility of mannose-containing glycoconjugates to fluorescent concanavalin A. In the absence of melanin, conidia showed a marked increase in fluorescence intensity as the age of culture increased. Using atomic force microscopy, we were unable to find rodlet-forming hydrophobins, molecules that can also affect conidial surface properties. In conclusion, the changes in surface properties and biochemical composition of the conidial wall with the age of culture highlight the process of conidial maturation. Mannose-containing glycoconjugates that are involved in immune recognition, are progressively masked by polymerization of melanin, an antioxidant that is commonly thought to allow fungal escape from the host immune defenses.
Genivaldo G.Z. Silva, Daniel A. Cuevas, Bas E. Dutilh and Robert A. Edwards (2014), "FOCUS: an alignment-free model to identify organisms in metagenomes using non-negative least squares", PeerJ 2: e425. Pubmed, PDF.
One of the major goals in metagenomics is to identify the organisms present in a microbial community from unannotated shotgun sequencing reads. Taxonomic profiling has valuable applications in biological and medical research, including disease diagnostics. Most currently available approaches do not scale well with increasing data volumes, which is important because both the number and lengths of the reads provided by sequencing platforms keep increasing. Here we introduce FOCUS, an agile composition based approach using non-negative least squares (NNLS) to report the organisms present in metagenomic samples and profile their abundances. FOCUS was tested with simulated and real metagenomes, and the results show that our approach accurately predicts the organisms present in microbial communities. FOCUS was implemented in Python. The source code and web-sever are freely available at http://edwards.sdsu.edu/FOCUS.
Marwa ElRakaiby, Bas E. Dutilh, Mariam R. Rizkallah, Annemarie Boleij, Jason N. Cole and Ramy K. Aziz (2014), "Pharmacomicrobiomics: the impact of human microbiome variations on systems pharmacology and personalized therapeutics", OMICS: A Journal of Integrative Biology 18: 402-414. Pubmed, PDF. Listed first in Mary Ann Liebert's Most Read Articles.
The Human Microbiome Project (HMP) is a global initiative undertaken to identify and characterize the collection of human-associated microorganisms at multiple anatomic sites (skin, mouth, nose, colon, vagina), and to determine how intraindividual and interindividual alterations in the microbiome influence human health, immunity, and different disease states. In this review article, we summarize the key findings and applications of the HMP that may impact pharmacology and personalized therapeutics. We propose a microbiome cloud model, reflecting the temporal and spatial uncertainty of defining an individual's microbiome composition, with examples of how intraindividual variations (such as age and mode of delivery) shape the microbiome structure. Additionally, we discuss how this microbiome cloud concept hinders the definition of a core human microbiome and the classification of individuals according to their biome types. Detailed examples are presented on microbiome changes related to colorectal cancer, antibiotic administration, and pharmacomicrobiomics, or drug-microbiome interactions, highlighting how an improved understanding of the human microbiome, and alterations thereof, may lead to the development of novel therapeutic agents, the modification of antibiotic policies and implementation, and improved health outcomes. Finally, the prospects of a collaborative computational microbiome research initiative in Africa are discussed.
Harold Tjalsma, Bas E. Dutilh, Annemarie Boleij, and Julian R. Marchesi (2014), "Colorectal cancer associated microbiota". In: Encyclopedia of Metagenomics III: Human Metagenomics. Eds. Sarah Highlander and Karen E. Nelson. Springer Reference.
Colorectal cancer (CRC) is one of the big killers in developed societies. More than one million new CRC cases are diagnosed and >600,000 patients die from this disease each year, making it the fourth most common cancer-associated cause of death. The genetic framework for this disease is formulated by the "adenoma-carcinoma sequence" based on the occurrence of driver mutations in crypt stem cells that render them immortal, and passenger mutations that accumulate as the tumor expands but which do not contribute directly to disease progression. Despite the fact that dietary and environmental factors (Western lifestyle), genetic background, and ethnicity have been associated with CRC risk, the exact molecular events that cause CRC driver mutations remain elusive. Important triggers may be derived from the dense and complex bacterial community of the gut that resides in close contact with the colonic mucosa and developing tumors. Recent clinical studies and experimental models have directly or indirectly linked the intestinal microbiota, or specific members thereof, to CRC progression.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Bas E. Dutilh, Lennart Backus, Sacha A.F.T. van Hijum and Harold Tjalsma (2013), "Screening metatranscriptomes for toxin genes as functional drivers of human colorectal cancer", Best Practice & Research: Clinical Gastroenterology 27: 85-99. Pubmed, PDF.
The colonic mucosa is in constant physical interaction with a dense and complex bacterial community that comprises health-promoting and pathogenic microbes. Here, we highlight important clinical studies and experimental models that have linked the intestinal microbiota to the development of colorectal cancer (CRC). Moreover, we use recently published metatranscriptome sequencing data to test whether potentially carcinogenic toxin genes exhibit higher expression levels in human CRC tissue compared to adjacent non-malignant mucosa. Our analyses show a large variation in expression of toxin(-related) genes from different species. Surprisingly, Enterobacterial toxins were among the highest expressed, while Enterobacteria were not among the most abundant species in these samples. Although we can differentiate on- and off-tumor sites based on toxin reads, the read depth profiles are quite similar and show only limited coverage of the toxin genes. Thus, extended metagenomic studies are needed to obtain a high-resolution picture of host-pathogen interactions during human CRC.
Bas E. Dutilh, Lennart Backus, Robert A. Edwards, Michiel Wels, Jumamurat R. Bayjanov and Sacha A.F.T. van Hijum (2013), "Explaining microbial phenotypes on a genomic scale: GWAS for microbes", Briefings in Functional Genomics 12: 366-380. Pubmed, PDF.
There is an increasing availability of complete or draft genome sequences for microbial organisms. These data form a potentially valuable resource for genotype-phenotype association and gene function prediction, provided that phenotypes are consistently annotated for all the sequenced strains. In this review, we address the requirements for successful gene-trait matching. We outline a basic protocol for microbial functional genomics, including genome assembly, annotation of genotypes (including SNPs, orthologous groups and prophages), data pre-processing, genotype-phenotype association, visualization and interpretation of results. The methodologies for association described herein can be applied to other data types, opening up possibilities to analyze transcriptome-phenotype associations, and correlate microbial population structure or activity, as measured by metagenomics, to environmental parameters.
Genivaldo G.Z. Silva, Bas E. Dutilh, T. David Matthews, Keri Elkins, Robert Schmieder, Elizabeth A. Dinsdale and Robert A. Edwards (2013), "Combining de novo and reference-guided assembly with Scaffold_builder". Source Code for Biology and Medicine 8: 23. Pubmed, PDF.
Genome sequencing has become routine, however genome assembly still remains a challenge despite the computational advances in the last decade. In particular, the abundance of repeat elements in genomes makes it difficult to assemble them into a single complete sequence. Identical repeats shorter than the average read length can generally be assembled without issue. However, longer repeats such as ribosomal RNA operons cannot be accurately assembled using existing tools. The application Scaffold_builder was designed to generate scaffolds - super contigs of sequences joined by N-bases - based on the homology to a closely related reference sequence. This is independent of mate-pair information and can be used complementarily for genome assembly, e.g. when mate-pairs are not available or have already been exploited. Scaffold_builder was evaluated using simulated pyrosequencing reads of the bacterial genomes Escherichia coli 042, Lactobacillus salivarius UCC118 and Salmonella enterica subsp. enterica serovar Typhi str. P-stx-12. Moreover, we sequenced two genomes from Salmonella enterica serovar Typhimurium LT2 G455 and Salmonella enterica serovar Typhimurium SDT1291 and show that Scaffold_builder decreases the number of contig sequences by 53% while more than doubling their average length. Scaffold_builder is written in Python and is available at http://edwards.sdsu.edu/scaffold_builder. A web-based implementation is additionally provided to allow users to submit a reference genome and a set of contigs to be scaffolded.
|
Genivaldo G. Z. Silva, Bas E. Dutilh, T. David Matthews, Keri Elkins, Elizabeth A. Dinsdale and Robert A. Edwards (2012), "Scaffold-builder for combining de novo and Reference-guided assembly", poster P48 at 10th Annual Rocky Mountain Bioinformatics Conference 2012, Snowmass, Colorado, USA. |

|
John L. Mokili, Bas E. Dutilh, Yan Wei Lim, Bradley S. Schneider, Travis Taylor, Matthew R. Haynes, David Metzgar, Christopher A. Myers, Patrick J. Blair, Bahador Nosrat, Nathan D. Wolfe and Forest Rohwer (2013), "Identification of a novel human papillomavirus by metagenomic analysis of samples from patients with febrile respiratory illness", PLoS ONE 8: e58404. Pubmed, PDF.
As part of a virus discovery investigation using a metagenomic approach, a highly divergent novel Human papillomavirus type was identified in pooled convenience nasal/oropharyngeal swab samples collected from patients with febrile respiratory illness. Phylogenetic analysis of the whole genome and the L1 gene reveals that the new HPV identified in this study clusters with previously described gamma papillomaviruses, sharing only 61.1% (whole genome) and 63.1% (L1) sequence identity with its closest relative in the Papillomavirus episteme (PAVE) database. This new virus was named HPV_SD2 pending official classification. The complete genome of HPV-SD2 is 7,299 bp long (36.3% G/C) and contains 7 open reading frames (L2, L1, E6, E7, E1, E2 and E4) and a non-coding long control region (LCR) between L1 and E6. The metagenomic procedures, coupled with the bioinformatic methods described herein are well suited to detect small circular genomes such as those of human papillomaviruses.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Bas E. Dutilh, Robert Schmieder, Jim Nulton, Ben Felts, Peter Salamon, Robert A. Edwards and John L. Mokili (2012), "Reference-independent comparative metagenomics using cross-assembly: crAss", Bioinformatics 28: 3225-3231. Pubmed, PDF. CrAss is now available as a Docker image thanks to the great efforts of OneStop Data Analysis. Thanks a lot!
Motivation: Metagenomes are often characterized by high levels of unknown sequences. Reads derived from known micro-organisms can easily be identified and analyzed using fast homology search algorithms and a suitable reference database, but the unknown sequences are often ignored in further analyses, biasing conclusions. Nevertheless, it is possible to use more data in a comparative metagenomic analysis by creating a cross-assembly of all reads, i.e. a single assembly of reads from different samples. Comparative metagenomics studies the inter-relationships between metagenomes from different samples. Using an assembly algorithm is a fast and intuitive way to link (partially) homologous reads without requiring a database of reference sequences. Results: Here, we introduce crAss, a novel bioinformatic tool that enables fast, simple analysis of cross-assembly files, yielding distances between all metagenomic sample pairs and an insightful image displaying the similarities. Availability and Implementation: crAss is available as a web server at http://edwards.sdsu.edu/crass/ and the Perl source code can be downloaded to run as a stand-alone, command line tool.
Bas E. Dutilh (2013), "Comparative metagenomics by cross-assembly", talk at 8th Benelux Bioinformatics Conference 2013, Brussels, Belgium.

Bas E. Dutilh, Robert Schmieder, Jim Nulton, Ben Felts, Peter Salamon, Robert A. Edwards and John L. Mokili (2013), "Comparative metagenomics by cross-assembly", 8th Benelux Bioinformatics Conference 2013. Brussels, Belgium. |

|
Simone M.T. Camps*, Bas E. Dutilh*, Maiken C. Arendrup, Antonius J.M.M. Rijs, Eveline Snelders, Martijn A. Huynen, Paul E. Verweij and Willem J.G. Melchers (2012), "Discovery of a hapE mutation that causes azole resistance in Aspergillus fumigatus through whole genome sequencing and sexual crossing", PLoS ONE 7: e50034. Pubmed, PDF. *Authors contributed equally. F1000 Must Read.
Azole compounds are the primary therapy for patients with diseases caused by Aspergillus fumigatus. However, prolonged treatment may cause resistance to develop, which is associated with treatment failure. The azole target cyp51A is a hotspot for mutations that confer phenotypic resistance, but in an increasing number of resistant isolates the underlying mechanism remains unknown. Here, we report the discovery of a novel resistance mechanism, caused by a mutation in the CCAAT-binding transcription factor complex subunit HapE. From one patient, four A. fumigatus isolates were serially collected. The last two isolates developed an azole resistant phenotype during prolonged azole therapy. Because the resistant isolates contained a wild type cyp51A gene and the isolates were isogenic, the complete genomes of the last susceptible isolate and the first resistant isolate (taken 17 weeks apart) were sequenced using Illumina technology to identify the resistance conferring mutation. By comparing the genome sequences to each other as well as to two A. fumigatus reference genomes, several potential non-synonymous mutations in protein-coding regions were identified, six of which could be confirmed by PCR and Sanger sequencing. Subsequent sexual crossing experiments showed that resistant progeny always contained a P88L substitution in HapE, while the presence of the other five mutations did not correlate with resistance in the progeny. Cloning the mutated hapE gene into the azole susceptible akuBKU80 strain showed that the HapE P88L mutation by itself could confer the resistant phenotype. This is the first time that whole genome sequencing and sexual crossing strategies have been used to find the genetic basis of a trait of interest in A. fumigatus. The discovery may help understand alternate pathways for azole resistance in A. fumigatus with implications for the molecular diagnosis of resistance and drug discovery.
Noriko Cassman*, Alejandra Prieto-Davó*, Kevin Walsh, Genivaldo G. Z. Silva, Florent Angly, Sajia Akhter, Katie Barott, Julia Busch, Tracey McDole, J. Matthew Haggerty, Dana Willner, Gadiel Alarcón, Osvaldo Ulloa, Edward F. DeLong, Bas E. Dutilh, Forest Rohwer and Elizabeth A. Dinsdale (2012), "Oxygen minimum zones harbor novel viral communities with low diversity", Environmental Microbiology 14: 3043-3065. Pubmed, PDF. *Authors contributed equally.
Oxygen minimum zones (OMZs) are oceanographic features that affect ocean productivity and biodiversity, and contribute to ocean nitrogen loss and greenhouse gas emissions. Here we describe the viral communities associated with the Eastern Tropical South Pacific (ETSP) OMZ off Iquique, Chile for the first time through abundance estimates and viral metagenomic analysis. The viral to microbial ratio (VMR) in the ETSP OMZ fluctuated in the oxycline and declined in the anoxic core to below one on several occasions. The number of viral genotypes (unique genomes as defined by sequence assembly) ranged from 2040 at the surface to 98 in the oxycline, which is the lowest viral diversity recorded to date in the ocean. Within the ETSP OMZ viromes, only 4.95 % of genotypes were shared between surface and anoxic core viromes using reciprocal BLASTn sequence comparison. ETSP virome comparison with surface marine viromes (Sargasso Sea, Gulf of Mexico, Kingman Reef, Chesapeake Bay) revealed a dissimilarity of ETSP OMZ viruses to those from other oceanic regions. From the 1.4 million non-redundant DNA sequences sampled within the altered oxygen conditions of the ETSP OMZ, more than 97.8 % were novel. Of the average 3.2 % of sequences that showed similarity to the SEED non-redundant database, phage sequences dominated the surface viromes, eukaryotic virus sequences dominated the oxycline viromes, and phage sequences dominated the anoxic core viromes. The viral community of the ETSP OMZ was characterized by fluctuations in abundance, taxa and diversity across the oxygen gradient. The ecological significance of these changes was difficult to predict, however, it appears that the reduction in oxygen coincides with an increased shedding of eukaryotic viruses in the oxycline, and a shift to unique viral genotypes in the anoxic core.
Bas E. Dutilh and Ramy K. Aziz (2012), "Genomes, metagenomes, and microbiomes: a new biology for a new millennium", New Life Sciences: Linking Science to Society, BioVision Alexandria 2012 Proceedings: 105-116. PDF (click here for the whole book).
Amaro E. Trindade-Silva, Cintia Rua, Genivaldo G. Z. Silva, Bas E. Dutilh, Ana Paula B. Moreira, Robert A. Edwards, Eduardo Hajdu, Gisele Lobo-Hajdu, Ana Tereza Vasconcelos, Roberto G. S. Berlinck, and Fabiano L. Thompson (2012), "Taxonomic and functional microbial signatures of the endemic marine sponge Arenosclera brasiliensis", PLoS ONE 7: e39905. Pubmed, PDF.
The endemic marine sponge Arenosclera brasiliensis (Porifera, Demospongiae, Haplosclerida) is a known source of secondary metabolites such as arenosclerins A-C. In the present study, we established the composition of the A. brasiliensis microbiome and the metabolic pathways associated with this community. We used 454 shotgun pyrosequencing to generate approximately 640,000 high-quality sponge-derived sequences (~150 Mb). Clustering analysis including sponge, seawater and twenty-three other metagenomes derived from marine animal microbiomes shows that A. brasiliensis contains a specific microbiome. Fourteen bacterial phyla (including Proteobacteria, Cyanobacteria, Actinobacteria, Bacteroidetes, Firmicutes and Chloroflexi) were consistently found in the A. brasiliensis metagenomes. The A. brasiliensis microbiome is enriched for Betaproteobacteria (e.g., Burkholderia) and Gammaproteobacteria (e.g., Pseudomonas and Alteromonas) compared with the surrounding planktonic microbial communities. Functional analysis based on Rapid Annotation using Subsystem Technology (RAST) indicated that the A. brasiliensis microbiome is enriched for sequences associated with membrane transport and one-carbon metabolism. In addition, there was an overrepresentation of sequences associated with aerobic and anaerobic metabolism as well as the synthesis and degradation of secondary metabolites. This study represents the first analysis of sponge-associated microbial communities via shotgun pyrosequencing, a strategy commonly applied in similar analyses in other marine invertebrate hosts, such as corals and algae. We demonstrate that A. brasiliensis has a unique microbiome that is distinct from that of the surrounding planktonic microbes and from other marine organisms, indicating a species-specific microbiome.
Harold Tjalsma, Annemarie Boleij, Julian R. Marchesi and Bas E. Dutilh (2012), "A Bacterial Driver-Passenger Model for Colorectal Cancer: Beyond the Usual Suspects", Nature Reviews Microbiology 10: 575-582. Pubmed, PDF.
Cancer has long been considered a genetic disease. However, accumulating evidence supports the involvement of infectious agents in the development of cancer, especially in those organs that are continuously exposed to microorganisms, such as the large intestine. Recent next-generation sequencing studies of the intestinal microbiota now offer an unprecedented view of the aetiology of sporadic colorectal cancer and have revealed that the microbiota associated with colorectal cancer contains bacterial species that differ in their temporal associations with developing tumours. Here, we propose a bacterial driver-passenger model for microbial involvement in the development of colorectal cancer and suggest that this model be incorporated into the genetic paradigm of cancer progression.
Annemarie Boleij, Bas E. Dutilh, Guus Kortman, Rian Roelofs, Coby M. Laarakkers, Udo F. Engelke and Harold Tjalsma (2012), "Bacterial Responses to a Simulated Colon Tumor Microenvironment", Molecular and Cellular Proteomics 11: 851-862. Pubmed, PDF.
One of the few bacteria that have been consistently linked to colorectal cancer (CRC) is the opportunistic pathogen Streptococcus gallolyticus. S. gallolyticus infections are generally regarded as an indicator for colonic malignancy, while the carriage rate of this bacterium in the healthy large intestine is relatively low. We speculated that the physiological changes accompanying the development of CRC might favor the colonization of this bacterium. To investigate whether colon tumor cells can support the survival of S. gallolyticus, S. gallolyticus was grown in spent medium of malignant colonocytes to simulate the altered metabolic conditions in the CRC microenvironment. These in vitro simulations indicated that S. gallolyticus had a significant growth advantage in these spent media, which was not observed for other intestinal bacteria. Under these conditions, bacterial responses were profiled by proteome analysis and metabolic shifts were analyzed by 1H-NMR-spectroscopy. In silico pathway analysis of the differentially expressed proteins and metabolite analysis indicated that this advantage resulted from the increased utilization of glucose, glucose derivates and alanine. Together, these data suggest that tumor cell metabolites facilitate the survival of S. gallolyticus, favoring its local outgrowth and providing a possible explanation for the specific association of S. gallolyticus with colonic malignancy.
Daniel R. Garza, Cristiane C. Thompson, Edvaldo C.B. Loureiro, Bas E. Dutilh, Davi T. Inada, Edivaldo C. Sousa Jr, Jedson F. Cardoso, Márcio R.T. Nunes, Clayton Pereira Silva de Lima, Rodrigo V.D. Silvestre, Keley N.B. Nunes, Elisabeth C.O. Santos, Robert A. Edwards, Ana C.P. Vicente and Lena L. Canto de Sá Morais (2012), "Genome-wide study of the defective sucrose fermenter strain of Vibrio cholerae from the Latin American cholera epidemic", PLoS ONE 7: e37283. Pubmed, PDF.
The 7th cholera pandemic reached Latin America in 1991, spreading from Peru to virtually all Latin American countries. During the late epidemic period, a strain that failed to ferment sucrose dominated cholera outbreaks in the Northern Brazilian Amazon region. In order to understand the genomic characteristics and the determinants of this altered sucrose fermenting phenotype, the genome of the strain IEC224 was sequenced. This paper reports a broad genomic study of this strain, showing its correlation with the major epidemic lineage. The potentially mobile genomic regions are shown to possess GC content deviation, and harbor the main V. cholerae virulence genes. A novel bioinformatic approach was applied in order to identify the putative functions of hypothetical proteins, and was compared with the automatic annotation by RAST. The genome of a large bacteriophage was found to be integrated to the IEC224's alanine aminopeptidase gene. The presence of this phage is shown to be a common characteristic of the El Tor strains from the Latin American epidemic, as well as its putative ancestor from Angola. The defective sucrose fermenting phenotype is shown to be due to a single nucleotide insertion in the V. cholerae sucrose-specific transportation gene. This frame-shift mutation truncated a membrane protein, altering its structural pore-like conformation. Further, the identification of a common bacteriophage reinforces both the monophyletic and African-Origin hypotheses for the main causative agent of the 1991 Latin America cholera epidemics.
Jack van de Vossenberg, Dagmar Woebken, Wouter J. Maalcke, Hans J.C.T. Wessels, Bas E. Dutilh, Boran Kartal, Eva M. Janssen-Megens, Guus Roeselers, Jia Yan, Daan Speth, Jolein Gloerich, Wim Geerts, Erwin van der Biezen, Wendy Pluk, Kees-Jan Françoijs, Lina Russ, Phyllis Lam, Stefanie A. Malfatti, Susannah Green Tringe, Suzanne C.M. Haaijer, Huub J.M. Op den Camp, Henk G. Stunnenberg, Rudi Amann, Marcel M.M. Kuypers and Mike S.M. Jetten (2012), "The metagenome of the marine anammox bacterium "Candidatus Scalindua profunda" illustrates the versatility of this globally important nitrogen cycle bacterium", Environmental Microbiology 15: 1275-1289. Pubmed, PDF.
Anaerobic ammonium oxidizing (anammox) bacteria are responsible for a significant portion of the loss of fixed nitrogen from the oceans, making them important players in the global nitrogen cycle. To date, marine anammox bacteria found in marine water columns and sediments worldwide belong almost exclusively to the "Candidatus Scalindua" species, but the molecular 5 basis of their metabolism and competitive fitness is presently unknown. We applied community sequencing of a marine anammox enrichment culture dominated by "Candidatus Scalindua profunda" to construct a genome assembly, which was subsequently used to analyze the most abundant gene transcripts and proteins. In the S. profunda assembly, 4756 genes were annotated, and only about half of them showed the highest identity to the only other anammox bacterium of which a metagenome assembly had been constructed so far, the fresh water "Candidatus Kuenenia stuttgartiensis". In total, 2016 genes of S. profunda could not be matched to the K. stuttgartiensis metagenome assembly at all, and a similar number of genes in K. stuttgartiensis could not be found in S. profunda. Most of these genes did not have a known function but 98 expressed genes could be attributed to oligopeptide transport, amino acid metabolism, use of organic acids, and electron transport. On the basis of the S. profunda metagenome, and environmental metagenome data, we observed pronounced differences in the gene organization and expression of important anammox enzymes, such as hydrazine synthase (HzsAB), nitrite reductase (NirS), and inorganic nitrogen transport proteins. Adaptations of Scalindua to the substrate limitation of the ocean may include highly expressed ammonium, nitrite and oligopeptide transport systems and pathways for the transport, oxidation, and assimilation of small organic compounds that may allow a more versatile lifestyle contributing to the competitive fitness of Scalindua in the marine realm.
Brigida T. Luckwu de Lucena, Genivaldo G.Z. Silva, Billy Manoel dos Santos, Graciela M. Dias, Gilda R. Amaral, Ana P. Moreira, Marcos A. de Morais Júnior, Bas E. Dutilh, Robert A. Edwards, Valdir Balbino, Cristiane C. Thompson and Fabiano L. Thompson (2012), "Genome sequence of the ethanol tolerant Lactobacillus vini strains LMG23202T and JP7.8.9", Journal of Bacteriology 194: 3018. Pubmed, PDF.
We report on the genome sequences of Lactobacillus vini LMG 23202T (DSM 20605) (isolated from fermenting grape musts in Spain) and the industrial strain L. vini JP7.8.9 (isolated from a bioethanol plant in northeast Brazil). All contigs were assembled using gsAssembler, and genes were predicted and annotated using Rapid Annotation using Subsystem Technology (RAST). The identified genome sequence of LMG 23202T had 2,201,333 bp, 37.6% G+C, and 1,833 genes, whereas the identified genome sequence of JP7.8.9 had 2,301,037 bp, 37.8% G+C, and 1,739 genes. The gene repertoire of the species L. vini offers promising opportunities for biotechnological applications.
John L. Mokili, Forest Rohwer and Bas E. Dutilh (2012), "Metagenomics and future perspectives in virus discovery", Current Opinion in Virology. 2: 63-77. Pubmed, DOI. Listed #17 in Elsevier Virology's most downloaded articles.
Monitoring the emergence and re-emergence of viral diseases with the goal of containing the spread of viral agents requires both adequate preparedness and quick response. Identifying the causative agent of a new epidemic is one of the most important steps for effective response to disease outbreaks. Traditionally, virus discovery required propagation of the virus in cell culture, a proven technique responsible for the identification of the vast majority of viruses known to date. However, many viruses cannot be easily propagated in cell culture, thus limiting our knowledge of viruses. Viral metagenomic analyses of environmental samples suggest that the field of virology has explored less than 1% of the extant viral diversity. In the last decade, the culture-independent and sequence-independent metagenomic approach has permitted the discovery of many viruses in a wide range of samples. Phylogenetically, some of these viruses are distantly related to previously discovered viruses. In addition, 60-99% of the sequences generated in different viral metagenomic studies are not homologous to known viruses. In this review, we discuss the advances in the area of viral metagenomics during the last decade and their relevance to virus discovery, clinical microbiology and public health. We discuss the potential of metagenomics for characterization of the normal viral population in a healthy community and identification of viruses that could pose a threat to humans through zoonosis. In addition, we propose a new model of the Koch's postulates named the 'Metagenomic Koch's Postulates'. Unlike the original Koch's postulates and the Molecular Koch's postulates as formulated by Falkow, the metagenomic Koch's postulates focus on the identification of metagenomic traits in disease cases. The metagenomic traits that can be traced after healthy individuals have been exposed to the source of the suspected pathogen.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Julian R. Marchesi, Bas E. Dutilh, Neil Hall, Wilbert H. M. Peters, Rian Roelofs, Annemarie Boleij and Harold Tjalsma (2011), "Towards the Human Colorectal Cancer Microbiome", PLoS ONE 6: e20447. Pubmed, PDF. F1000 Recommended.
Multiple factors drive the progression from healthy mucosa towards sporadic colorectal carcinomas and accumulating evidence associates intestinal bacteria with disease initiation and progression. Therefore, the aim of this study was to provide a first high-resolution map of colonic dysbiosis that is associated with human colorectal cancer (CRC). To this purpose, the microbiomes colonizing colon tumor tissue and adjacent non-malignant mucosa were compared by deep rRNA sequencing. The results revealed striking differences in microbial colonization patterns between these two sites. Although inter-individual colonization in CRC patients was variable, tumors consistently formed a niche for Coriobacteria and other proposed probiotic bacterial species, while potentially pathogenic Enterobacteria were underrepresented in tumor tissue. As the intestinal microbiota is generally stable during adult life, these findings suggest that CRC-associated physiological and metabolic changes recruit tumor-foraging commensal-like bacteria. These microbes thus have an apparent competitive advantage in the tumor microenvironment and thereby seem to replace pathogenic bacteria that may be implicated in CRC etiology. This first glimpse of the CRC microbiome provides an important step towards full understanding of the dynamic interplay between intestinal microbial ecology and sporadic CRC, which may provide important leads towards novel microbiome-related diagnostic tools and therapeutic interventions.
|
Bas E. Dutilh, Julian R Marchesi, Annemarie Boleij and Harold Tjalsma (2012), "Towards the human colorectal cancer microbiome", poster at MetaHIT International Human Microbiome Congress, Paris, France. |

|
Maartje A.H.J. van Kessel, Bas E. Dutilh, Kornelia Neveling, Michael P. Kwint, Joris A. Veltman, Gert Flik, Mike S.M. Jetten, Peter H.M. Klaren and Huub J.M. Op den Camp (2011), "Pyrosequencing of 16S rRNA gene amplicons to study the microbiota in the gastrointestinal tract of carp (Cyprinus carpio L.)", AMB Express 1: 41. Pubmed, PDF.
The microbes in the gastrointestinal (GI) tract are of high importance for the health of the host. In this study, Roche 454 pyrosequencing was applied to a pooled set of different 16S rRNA gene amplicons obtained from GI content of common carp (Cyprinus carpio) to make an inventory of the diversity of the microbiota in the GI tract. Compared to other studies, our culture-independent investigation reveals an impressive diversity of the microbial flora of the carp GI tract. The major group of obtained sequences belonged to the phylum Fusobacteria. Bacteroidetes, Planctomycetes and Gammaproteobacteria were other well represented groups of micro-organisms. Verrucomicrobiae, Clostridia and Bacilli (the latter two belonging to the phylum Firmicutes) had fewer representatives among the analyzed sequences. Many of these bacteria might be of high physiological relevance for carp as these groups have been implicated in vitamin production, nitrogen cycling and (cellulose) fermentation.
Cristiane C. Thompson, Michel Abanto Marin, Graciela Maria Dias, Bas E. Dutilh, Rob Edwards, Tetsuya Iida and Fabiano L. Thompson (2011), "Genome sequence of the human pathogen Vibrio cholerae Amazonia", Journal of Bacteriology 193: 5877-5878. Pubmed, PDF.
Vibrio cholerae O1 Amazonia is a pathogen that was isolated from cholera like diarrhea cases in, at least, two countries, Brazil and Ghana. It belongs to a distinct profile by MLSA. The genomic analysis revealed that it contains the Vibrio pathogenicity island-2 and a set of genes related with pathogenesis and fitness, as the type VI secretion system, present in choleragenic V. cholerae strains.
Bas E. Dutilh, Rasa Jurgelenaite, Radek Szklarczyk, Sacha A.F.T. van Hijum, Harry R. Harhangi, Markus Schmid, Bart de Wild, Kees-Jan Françoijs, Hendrik G. Stunnenberg, Marc Strous, Mike S.M. Jetten, Huub J.M. Op den Camp and Martijn A. Huynen (2011), "FACIL: fast and accurate genetic code inference and logo", Bioinformatics 27: 1929-1933. Pubmed, PDF.
Motivation: The intensification of environmental DNA sequencing will increasingly unveil uncharacterized species with potential alternative genetic codes. A total of 0.65% of the DNA sequences currently in Genbank encode their proteins with a variant genetic code, and these exceptions occur in many unrelated taxa. Results: We introduce FACIL, a fast and reliable tool to evaluate nucleic acid sequences for their genetic code that detects alternative codes even in species distantly related to known organisms. To illustrate this, we apply FACIL to a set of mitochondrial genomic contigs of Globobulimina pseudospinescens. This foraminifer does not have any sequenced close relatives in the databases, yet we infer its alternative genetic code with high confidence values. Results are intuitively visualized in a Genetic Code Logo. Availability and Implementation: FACIL is available as a web-based service at http://www.cmbi.ru.nl/FACIL/ and as a stand-alone program.
Bas E. Dutilh (2011), "FACIL: fast and accurate genetic code inference and logo", talk at San Diego Microbiology Group All Day Meeting 2011, San Diego, California, USA.
|
Bas E. Dutilh, Rasa Jurgelenaite, Radek Szklarczyk, Sacha A.F.T. van Hijum, Harry R. Harhangi, Markus Schmid, Bart de Wild, Kees-Jan Françoijs, Hendrik G. Stunnenberg, Marc Strous, Mike S.M. Jetten, Huub J.M. Op den Camp and Martijn A. Huynen (2011), "FACIL: fast and accurate genetic code inference and logo", poster at San Diego Microbiology Group All Day Meeting 2011, San Diego, California, USA. |

|
Nardy Kip, Bas E. Dutilh, Yao Pan, Levente Bodrossy, Kornelia Neveling, Michael P. Kwint, Mike S.M. Jetten and Huub J.M. Op den Camp (2011), "Ultra-deep pyrosequencing of pmoA amplicons confirms prevalence of Methylomonas and Methylocystis in Sphagnum mosses from a Dutch peat bog", Environmental Microbiology Reports 3: no. doi: 10.1111/j.1758-2229.2011.00260.x. PDF.
Sphagnum peatlands are important ecosystems in the methane cycle. Methanotrophs in these ecosystems have been shown to reduce methane emissions and provide additional carbon to Sphagnum mosses. However, little is known about the diversity and identity of the methanotrophs present in and on Sphagnum mosses in these peatlands. In this study, we applied a pmoA microarray and high-throughput 454 pyrosequencing to pmoA PCR products obtained from total DNA from Sphagnum mosses from a Dutch peat bog to investigate the presence of methanotrophs and to compare the two different methods. Both techniques showed comparable results and revealed an abundance of Methylomonas and Methylocystis species in the Sphagnum mosses. The advantage of the microarray analysis is that it is fast and cost-effective, especially when many samples have to be screened. Pyrosequencing is superior in providing pmoA sequences of many unknown or uncultivated methanotrophs present in the Sphagnum mosses and, thus, provided much more detailed and quantitative insight into the microbial diversity.
Harold Tjalsma*, Coby M.M. Laarakkers*, Rachel P.S. van Swelm, Milan Theurl, Igor Theurl, Erwin H. Kemna, Yuri E.M. van der Burgt, Hanka Venselaar, Bas E. Dutilh, Frans G.M. Russel, Günter Weiss, Rosalinde Masereeuw, Robert E. Fleming, Dorine W. Swinkels (2011), "Mass Spectrometry Analysis of Hepcidin Peptides in Experimental Mouse Models", PLoS ONE 6: e16762. Pubmed, PDF. *Authors contributed equally.
Background The mouse is a valuable model for unravelling the role of hepcidin in iron homeostasis. Here, we aimed to assess mouse hepcidin-1 (Hep-1) and -2 (Hep-2) peptide levels in serum and urine by a novel mass spectrometry (MS)-based approach. Methods We used time-of-flight (TOF) MS to determine Hep-1 and -2 levels and Fourier transform ion cyclotron resonance (FTICR) and tandem-MS for hepcidin identifications. The method was biologically validated by hepcidin assessment in: i) 3 mouse strains (C57Bl/6; DBA/2 and BABL/c) upon stimulation with intravenous iron and LPS, ii) homozygous Hfe knock out, homozygous transferrin receptor 2 (Y245X) mutated mice and double affected mice, and iii) mice treated with a sublethal hepatotoxic dose of paracetamol. Results Hep-1 detection was restricted to serum, while Hep-2 was only found in urine and consisted of several isoforms. Elevations in serum Hep-1 and urine Hep-2 upon intravenous iron or LPS were only moderate and varied considerably between mouse strains. Serum Hep-1 was decreased in all three hemochromatosis models and lowest in the double affected mouse. Serum Hep-1 levels correlated with liver hepcidin-1 gene expression, while acute liver damage by paracetamol depleted Hep-1 from serum. Furthermore, serum Hep-1 appeared to be an excellent indicator of splenic iron accumulation. Conclusion Hep-1 and Hep-2 peptide responses in experimental mouse agree with the known biology of hepcidin mRNA regulators, and their measurement can now be implemented in iron-related experimental mouse models to provide novel insights in post-transcriptional regulation, hepcidin function, and kinetics.
Rob M. de Graaf*, Guenola Ricard*, Theo A. van Alen, Isabel Duarte, Bas E. Dutilh, Carola Burgtorf, Jan W.P. Kuiper, Georg W.M. van der Staay, Aloysius G.M. Tielens, Martijn A. Huynen and Johannes H.P. Hackstein (2011), "The organellar genome and metabolic potential of the hydrogen-producing mitochondrion of Nyctotherus ovalis", Molecular Biology and Evolution 28: 2379-2391. Pubmed, PDF. *Authors contributed equally.
It is generally accepted that hydrogenosomes (hydrogen-producing organelles) evolved from a mitochondrial ancestor. However, until recently, only indirect evidence for this hypothesis was available. Here we present the almost complete genome of the hydrogen-producing mitochondrion of the anaerobic ciliate Nyctotherus ovalis and show that, except for the notable absence of genes encoding electron-transport chain components of Complexes III, IV and V, it has a gene content similar to the mitochondrial genomes of aerobic ciliates. Analysis of the genome of the hydrogen-producing mitochondrion, in combination with that of more than 9,000 gDNA and cDNA sequences, allows a preliminary reconstruction of the organellar metabolism. The sequence data indicate that N. ovalis possesses hydrogen-producing mitochondria that have a truncated, two step (Complex I and II) electron-transport chain that uses fumarate as electron acceptor. In addition, components of an extensive protein network for the metabolism of amino-acids, defense against oxidative stress, mitochondrial protein synthesis, mitochondrial protein import and processing, and transport of metabolites across the mitochondrial membrane were identified. Genes for MPV17 and ACN9, two hypothetical proteins linked to mitochondrial disease in humans, were also found. The inferred metabolism is remarkably similar to the organellar metabolism of the phylogenetically distant anaerobic Stramenopile Blastocystis. Notably, the Blastocystis organelle and that of the related flagellate Proteromonas lacertae also lacks genes encoding components of Complexes III, IV and V. Thus, our data show that the hydrogenosomes of N. ovalis are highly specialized, hydrogen-producing mitochondria.
Joyce Emons, Bas E. Dutilh, Eva Decker, Heide Pirzer, Carsten Sticht, Norbert Gretz, Gudrun Rappold, Ewen R. Cameron, James C. Neil, Gary S. Stein, Andre J. van Wijnen, Jan Maarten Wit, Janine N. Post, Marcel Karperien (2011), "Genome wide screening in human growth plates during puberty in one patient suggests a role for RUNX2 in epiphyseal maturation", Journal of Endocrinology 209: 245-254. Pubmed, PDF.
In late puberty, estrogen decelerates bone growth by stimulating growth plate maturation. Here, we studied the mechanism of estrogen action using two pubertal growth plate specimens of one girl at Tanner stage B2 and Tanner stage B3. Histological analysis showed that progression of puberty coincided with characteristic morphological changes; a decrease in total growth plate height (p=0.002), height of the individual zones (p<0.001) and an increase in intercolumnar space (p<0.001). Microarray analysis of the specimens identified 394 genes (72% upregulated, 28% downregulated) that changed with the progression of puberty. Overall changes in gene expression were small (average 1.38-fold upregulated and 1.36-fold downregulated genes). The 394 genes mapped to 13 significantly changing pathways (p<0.05) associated with growth plate maturation (e.g., extracellular matrix, cell cycle and cell death). We next scanned the upstream promoter regions of the 394 genes for the presence of evolutionarily conserved binding sites for transcription factors implicated in growth plate maturation such as Estrogen Receptor, Androgen Receptor, Elk1, Stat5b, CREB and RUNX2. High quality motif sites for RUNX2 (87 genes), Elk1 (43 genes) and Stat5b (31 genes), but not estrogen receptor, were evolutionarily conserved, indicating their functional relevance across primates. Moreover, we show that some of these sites are direct target genes of these transcription factors as shown by ChIP assays.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Evelyn N. Kouwenhoven*, Simon J. van Heeringen*, Juan J. Tena*, Martin Oti, Bas E. Dutilh, M. Eva Alonso, Elisa de la Calle-Mustienes, Leonie Smeenk, Tuula Rinne, Lilian Parsaulian, Emine Bolat, Rasa Jurgelenaite, Martijn A. Huynen, Alexander Hoischen, Joris A. Veltman, Han G. Brunner, Tony Roscioli, Emily Oates, Meredith Wilson, Miguel Manzanares, José Luis Gómez-Skarmeta, Hendrik G. Stunnenberg, Marion Lohrum, Hans van Bokhoven and Huiqing Zhou (2010), "Genome-wide profiling of p63 DNA-binding sites identifies an element that regulates gene expression during limb development in the 7q21 SHFM1 locus", PLoS Genetics 6: e1001065. Pubmed, PDF. *Authors contributed equally.
Heterozygous mutations in p63 are associated with split hand/foot malformations (SHFM), orofacial clefting and ectodermal abnormalities. Elucidation of the p63 gene network that includes target genes and regulatory elements may reveal new genes for other malformation disorders. We performed genome-wide DNA-binding profiling by chromatin immunoprecipitation (ChIP) followed by deep sequencing (ChIP-seq) in primary human keratinocytes, and identified potential target genes and regulatory elements controlled by p63. We show that p63 binds to an enhancer element in the SHFM1 locus on chromosome 7q and that this element controls expression of DLX6 and possibly DLX5, both of which are important for limb development. A unique microdeletion including this enhancer element but not the DLX5/DLX6 genes was identified in a patient with SHFM. Our study strongly indicates disruption of a non-coding cis-regulatory element located more than 250 kb from the DLX5/DLX6 genes as a novel disease mechanism in SHFM1. These data provide a proof-of-concept that the catalogue of p63 binding sites identified in this study may be of relevance to the studies of SHFM and other congenital malformations that resemble the p63-associated phenotypes.
Bas E. Dutilh (2010). "Deconstructing the super-organism: detecting metabolic differentiation by compartmentalizing metagenomes", Veni award, NWO.
This Veni award from the Netherlands Organization for Scientific Research (NWO) enables me to do 3 years of independent research. Click here for the results. I will interpret the functionality of metagenomes at the level of individual micro-organisms. The award has been highlighted by Gezondheidskrant.nl, Medicalfacts.nl.
Katharina F. Ettwig*, Margaret K. Butler*, Denis Le Paslier, Eric Pelletier, Sophie Mangenot, Marcel M.M. Kuypers, Frank Schreiber, Bas E. Dutilh, Johannes Zedelius, Dirk de Beer, Jolein Gloerich, Hans J.C.T. Wessels, Theo A. van Alen, Francisca Luesken, Ming L. Wu, Katinka T. van de Pas-Schoonen, Huub J.M. Op den Camp, Eva M. Janssen-Megens, Kees-Jan Francoijs, Henk Stunnenberg, Jean Weissenbach, Mike S.M. Jetten and Marc Strous (2010), "Nitrite-driven anaerobic methane oxidation by oxygenic bacteria", Nature 464: 543-548. Pubmed, PDF, F1000 Exceptional. *Authors contributed equally.
Only three biological pathways are known to produce oxygen: photosynthesis, chlorate respiration and the detoxification of reactive oxygen species. Here we present evidence for a fourth pathway, possibly of considerable geochemical and evolutionary importance. The pathway was discovered after metagenomic sequencing of an enrichment culture that couples anaerobic oxidation of methane with the reduction of nitrite to dinitrogen. The complete genome of the dominant bacterium, named 'Candidatus Methylomirabilis oxyfera', was assembled. This apparently anaerobic, denitrifying bacterium encoded, transcribed and expressed the well-established aerobic pathway for methane oxidation, whereas it lacked known genes for dinitrogen production. Subsequent isotopic labelling indicated that 'M. oxyfera' bypassed the denitrification intermediate nitrous oxide by the conversion of two nitric oxide molecules to dinitrogen and oxygen, which was used to oxidize methane. These results extend our understanding of hydrocarbon degradation under anoxic conditions and explain the biochemical mechanism of a poorly understood freshwater methane sink. Because nitrogen oxides were already present on early Earth, our finding opens up the possibility that oxygen was available to microbial metabolism before the evolution of oxygenic photosynthesis.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Rob M. de Graaf, Theo A. van Alen, Bas E. Dutilh, Jan W.P. Kuiper, Hanneke J.A.A. van Zoggel, Minh Bao Huynh, Hans-Dieter Görtz, Martijn A. Huynen and Johannes H.P. Hackstein (2009), "The mitochondrial genomes of the ciliates Euplotes minuta and Euplotes crassus", BMC Genomics 10: 514. PDF, Pubmed.
Background There are thousands of very diverse ciliates species from which only a handful mitochondrial genomes have been studied so far. These genomes are rather similar because the ciliates analysed (Tetrahymena spp. and Paramecium aurelia) are closely related. Here we study the mitochondrial genomes of the hypotrichous ciliates Euplotes minuta and Euplotes crassus. These ciliates are only distantly related to Tetrahymena spp. and Paramecium aurelia, but more closely related to Nyctotherus ovalis, which possesses a hydrogenosomal (mitochondrial) genome. Results The linear mitochondrial genomes of the hypotrichous ciliates Euplotes minuta and Euplotes crassus were sequenced and compared with the mitochondrial genomes of several Tetrahymena species, Paramecium aurelia and the partially sequenced mitochondrial genome of the anaerobic ciliate Nyctotherus ovalis. This study reports new features such as long 5'gene extensions of several mitochondrial genes, extremely long cox1 and cox2 open reading frames and a large repeat in the middle of the linear mitochondrial genome. The repeat separates the open reading frames into two blocks, each having a single direction of transcription, from the repeat towards the ends of the chromosome. Although the Euplotes mitochondrial gene content is almost identical to Paramecium and Tetrahymena, the order of the genes is completely different. In contrast, the 33273 bp (excluding the repeat region) piece of the mitochondrial genome that has been sequenced in both Euplotes species exhibits no difference in gene order. Unexpectedly, many of the mitochondrial genes of E. minuta encoding ribosomal proteins possess N-terminal extensions that are similar to mitochondrial targeting signals. Conclusions The mitochondrial genomes of the hypotrichous ciliates Euplotes minuta and Euplotes crassus are rather different from the previously studied genomes. Many genes are extended in size compared to mitochondrial genes from other sources.
Bas E. Dutilh, Martijn A. Huynen and Marc Strous (2009), "Increasing the coverage of a metapopulation consensus genome by iterative read mapping and assembly", Bioinformatics 25: 2878-2881, PDF, Pubmed.
Motivation Most microbial species can not be cultured in the lab. Metagenomic sequencing may still yield a complete genome if the sequenced community is enriched and the sequencing coverage is high. However, the complexity in a natural population may cause the enrichment culture to contain multiple related strains. This diversity can confound existing strict assembly programs and lead to a fragmented assembly, which is unnecessary if we have a related reference genome available that can function as a scaffold. Results Here, we map short metagenomic sequencing reads from a population of strains to a related reference genome, and compose a genome that captures the consensus of the population's sequences. We show that by iteration of the mapping and assembly procedure, the coverage increases while the similarity with the reference genome decreases. This indicates that the assembly becomes less dependent on the reference genome and approaches the consensus genome of the multi-strain population.
Bas E. Dutilh, Martijn A. Huynen, Jolein Gloerich and Marc Strous (2011), "Iterative Read Mapping and Assembly Allows the Use of a More Distant Reference in Metagenome Assembly". In: Handbook of Molecular Microbial Ecology I: Metagenomics and Complementary Approaches. Ed. Frans J. de Bruijn. Wiley-Blackwell.
Most microbial species can not be cultured in the laboratory. Metagenomic sequencing may still yield a complete genome if the sequenced community is enriched and the sequencing coverage is high. However, the complexity in a natural population may cause the enrichment culture to contain multiple related strains. Moreover, it is not uncommon that these strains represent a quasispecies that is relatively distantly related to the closest available reference genome. These matters can confound existing strict assembly programs and lead to a fragmented assembly, which is unnecessary if we have a related reference genome available that can function as a scaffold, and if we use this scaffold loosely. We show that by iteratively mapping short metagenomic sequencing reads from a population of strains to a related reference genome, we can create a genome that captures the consensus of the population's sequences. Iteration allows us to map more of the reads, leading to a higher coverage and depth of the assembled consensus genome. At the same time, the similarity with the reference genome decreases. This indicates that the assembly becomes less dependent on the reference genome and approaches the consensus genome of the multi-strain population. Thus, by exploiting the homology offered by a reference genome in combination with permissive, iterative read mapping, we get a better view of both the consensus genome sequence of the quasispecies present in the sample and of the sequence diversity between the strains.

Bas E. Dutilh (2010), "Iterative read mapping and assembly allows the use of a more distant reference in metagenome assembly", talk at Bio-IT World Conference and Expo 2010, Hannover, Germany.
Bas E. Dutilh (2010), "Iterative read mapping and assembly allows the use of a more distant reference in metagenome assembly", talk at Genomics Automation Europe 2010, Dublin, Ireland.
Bas E. Dutilh (2010), "Increasing the coverage of a metapopulation consensus genome by iterative read mapping and assembly", talk at NBIC Conference 2010, Lunteren, The Netherlands.
Bas E. Dutilh, Martijn A. Huynen and Marc Strous (2009), "Increasing the coverage of a metapopulation consensus genome by iterative read mapping and assembly", talk at Next Generation Sequencing and Algorithms for Short Read Analysis SIG, ISMB/ECCB 2009, Stockholm, Sweden.
|
Bas E. Dutilh, Martijn A. Huynen, Jolein Gloerich and Marc Strous (2010), "Iterative read mapping and assembly allows the use of a more distant reference in metagenome assembly", poster at ECCB 2010, Ghent, Belgium. Bas E. Dutilh, Martijn A. Huynen, Jolein Gloerich and Marc Strous (2010), "Iterative read mapping and assembly allows the use of a more distant reference in metagenome assembly", poster at NBIC Conference 2010, Lunteren, The Netherlands. |
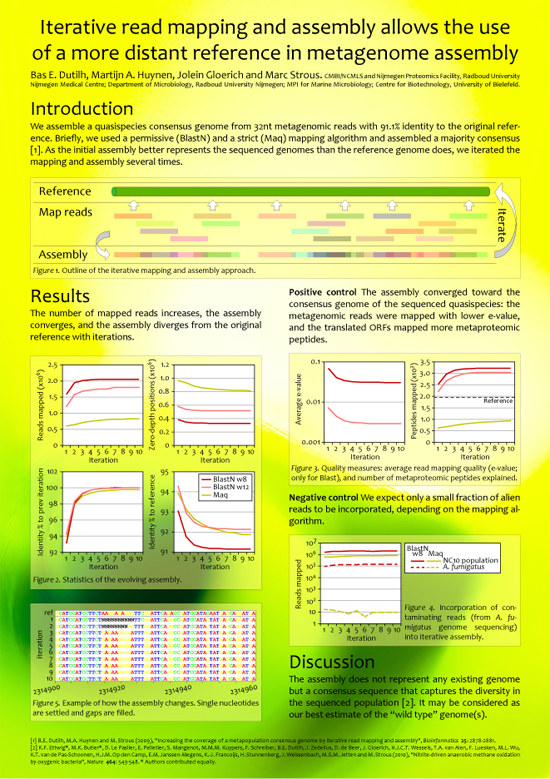
|
Richard A. Notebaart*, Philip R. Kensche*, Martijn A. Huynen and Bas E. Dutilh (2009), "Asymmetric relationships between proteins shape genome evolution", Genome Biology 10: R19. Pubmed, PDF. *Authors contributed equally.
Background The relationships between proteins are often asymmetric: one protein (A) depends for its function on another protein (B), but the second protein does not depend on the first. For example, in regulatory interactions, the regulator's function depends on the availability of its target, but the target can often function without the regulator. Other examples are metabolic networks, in which there are multiple pathways that converge into one central pathway. The enzymes in the converging pathways depend on the enzymes in the central pathway, but the enzymes in the latter do not depend on any specific enzyme in the converging pathways. Asymmetric relations are analogous to the "if->then" logical relation where A implies B, but B does not imply A (A->B). Results We show that the majority of relationships between enzymes in metabolic flux models of metabolism in Escherichia coli and Saccharomyces cerevisiae are asymmetric. We show furthermore that these asymmetric relationships are reflected in the expression of the genes encoding those enzymes, the effect of gene knockouts and the evolution of genomes. From the asymmetric relative dependency, one would expect that the gene that is relatively independent (B), can occur without the other, dependent gene (A), but not the reverse. Indeed, when only one gene of an A->B pair is expressed, is essential, is present in a genome, is gained in evolutionary history without the other, or is present after a loss of one of the two, it tends to be the independent gene (B). This bias is strongest for genes encoding proteins whose asymmetric relationship is evolutionarily conserved. Conclusions The asymmetric relations between proteins that arise from the system properties of metabolic networks affect gene expression, the relative effect of gene knockouts and genome evolution in a predictable manner.
|
Richard A. Notebaart*, Philip R. Kensche*, Martijn A. Huynen and Bas E. Dutilh (2009),
"Asymmetric relationships between proteins shape genome evolution",
poster at ISMB/ECCB 2009, Stockholm, Sweden.
Philip R. Kensche*, Richard A. Notebaart*, Martijn A. Huynen and Bas E. Dutilh (2008),
"Asymmetric relationships between proteins shape genome evolution",
poster at Benelux Bioinformatics Conference 2008, Maastricht, The Netherlands.
|

|
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Guénola Ricard, Rob M. de Graaf, Bas E. Dutilh, Isabel Duarte, Theo A. van Alen, Angela H.A.M. van Hoek, Brigitte Boxma, Georg W.M. van der Staay, Seung Yeo Moon van der Staay, Wei-Jen Chang, Laura F. Landweber, Johannes H.P. Hackstein and Martijn A. Huynen (2008), "Macronuclear genome structure of the ciliate Nyctotherus ovalis: Single-gene chromosomes and tiny introns", BMC Genomics 9: 587. Pubmed, PDF.
Background Nyctotherus ovalis is a single-celled eukaryote that has hydrogen-producing mitochondria and lives in the hindgut of cockroaches. Like all members of the ciliate taxon, it has two types of nuclei, a micronucleus and a macronucleus. N. ovalis generates its macronuclear chromosomes by forming polytene chromosomes that subsequently develop into macronuclear chromosomes by DNA elimination and rearrangement. Results We examined the structure of these gene-sized macronuclear chromosomes in N. ovalis. We determined the telomeres, subtelomeric regions, UTRs, coding regions and introns by sequencing a large set of macronuclear DNA sequences (4,242) and cDNAs (5,484) and comparing them with each other. The telomeres consist of repeats CCC(AAAACCCC)n, similar to those in spirotrichous ciliates such as Euplotes, Sterkiella (Oxytricha) and Stylonychia. Per sequenced chromosome we found evidence for either a single protein-coding gene, a single tRNA, or the complete ribosomal RNAs cluster. Hence the chromosomes appear to encode single transcripts. In the short subtelomeric regions we identified a few over-represented motifs that could be involved in gene regulation, but there is no consensus polyadenylation site. The introns are short (21-29 nucleotides), and a significant fraction (1/3) of the tiny introns is conserved in the distantly related ciliate Paramecium tetraurelia. As has been observed in P. tetraurelia, the N. ovalis introns tend to contain in-frame stop codons or have a length that is not dividable by three. This pattern causes premature termination of mRNA translation in the event of intron retention, and potentially degradation of unspliced mRNAs by the nonsense-mediated mRNA decay pathway. Conclusions The combination of short leaders, tiny introns and single genes leads to very minimal macronuclear chromosomes. The smallest we identified contained only 150 nucleotides.
Bas E. Dutilh, Berend Snel, Thijs J.G. Ettema and Martijn A. Huynen (2008), "Signature genes as a phylogenomic tool", Molecular Biology and Evolution 25: 1659-1667. Pubmed, PDF.
Gene content has been shown to contain a strong phylogenetic signal, yet its usage for phylogenetic
questions is hampered by horizontal gene transfer and parallel gene loss, and until now required
completely sequenced genomes.
Here, we introduce an approach that allows the phylogenetic signal in gene content to be applied to
any set of sequences, using signature genes for phylogenetic classification.
The hundreds of publicly available genomes allow us to identify signature genes at various taxonomic
depths, and we show how the presence of signature genes in an unspecified sample can be used to
characterize its taxonomic composition.
We identify 8,362 signature genes specific for 112 prokaryotic taxa. We show that these signature
genes can be used to address phylogenetic questions on the basis of gene content in cases where
classic gene content or sequence analyses provide an ambiguous answer, such as for Nanoarchaeum
equitans, and even in cases where complete genomes are not available, such as for metagenomics data.
Cross-validation experiments leaving out up to 30% of the species show that ~92% of the signature
genes correctly place the species in a related clade. Analyses of metagenomics data sets with the
signature gene approach are in good agreement with the previously reported species distributions
based on phylogenetic analysis of marker genes.
Summarising, signature genes can complement traditional sequence based methods in addressing
taxonomic questions.
Bas E. Dutilh, Ying He, Maarten L. Hekkelman and Martijn A. Huynen (2008), "Signature, a web server for taxonomic characterization of sequence samples using signature genes", Nucleic Acids Res, 36 (Web Server Issue): W470-W474. Pubmed, PDF.
Signature genes are genes that are unique to a taxonomic clade and are common within it. They contain a wealth of information about clade-specific 15 processes and hold a strong evolutionary signal that can be used to phylogenetically characterize a set of sequences, such as a metagenomics sample. As signature genes are based on gene content, they provide a means to assess the taxonomic origin 20 of a sequence sample that is complementary to sequence-based analyses. Here, we introduce Signature (http://www.cmbi.ru.nl/signature), a web server that identifies the signature genes in a set of query sequences, and therewith 25 phylogenetically characterizes it. The server produces a list of taxonomic clades that share signature genes with the set of query sequences, along with an insightful image of the tree of life, in which the clades are color coded based on the number of 30 signature genes present. This allows the user to quickly see from which part(s) of the taxonomy the query sequences likely originate.

Signature genes are genes with a common ancestor, that are specific for a clade in the Tree of Life, and can be used to address phylogenetic or functional questions. Signature allows you to find out whether your sequence is a signature for any clade, and places the signature OGs in the context of the tre of life. Your initial input is first assigned to orthologous groups (OGs), or you can choose to skip the OG assignment step and enter OG identifiers directly. The distribution of these OGs is assessed in a default or custom tree of life, and finally Signature outputs all the clades that share signature OGs with your query.

The Bioscience Technology article Characterizing The Tree Of Life includes an interview with me about Signature.
|
Bas E. Dutilh, Berend Snel, Thijs J.G. Ettema, Ying He, Maarten L. Hekkelman and Martijn A. Huynen (2008), "Signature genes as a phylogenomic tool", poster at ISMB/ECCB 2009, Stockholm, Sweden.
Bas E. Dutilh, Berend Snel, Thijs J.G. Ettema, Ying He, Maarten L. Hekkelman and Martijn A. Huynen (2008),
"Signature genes as a phylogenomic tool",
poster at Society for Bioinformatics in Northern Europe Conference 2008, Warszawa, Poland.
|
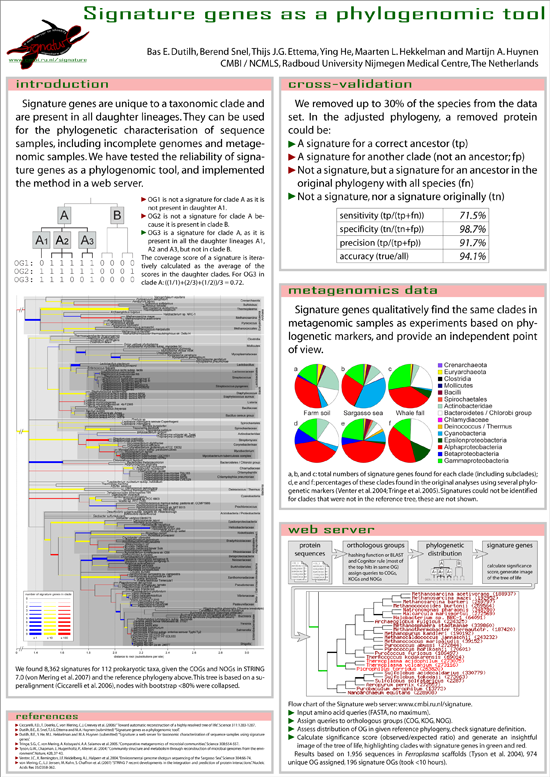
|
Philip R. Kensche, Martin Oti, Bas E. Dutilh and Martijn A. Huynen (2008), "Conservation of divergent transcription in fungi", Trends in Genetics 24: 207-211. PubMed, PDF.
The comparison of fully sequenced genomes enables the study of selective constraints that determine genome organisation. We show that, in fungi, adjacent divergently transcribed (<-->) genes are more conserved in orientation than convergent (-><-) or co-oriented (->->) gene pairs. Furthermore, the time divergent orientation of two genes is conserved correlates with the degree of their co-expression and with the likelihood of them being functionally related. The functional interactions of the proteins encoded by the conserved divergent gene pairs indicate a potential for protein function prediction in eukaryotes.
|
Philip R. Kensche, Martin Oti, Bas E. Dutilh and Martijn A. Huynen (2008),
"Conservation of Divergent Transcription in Fungi",
poster at Society for Bioinformatics in Northern Europe Conference 2008, Warszawa, Poland.
|
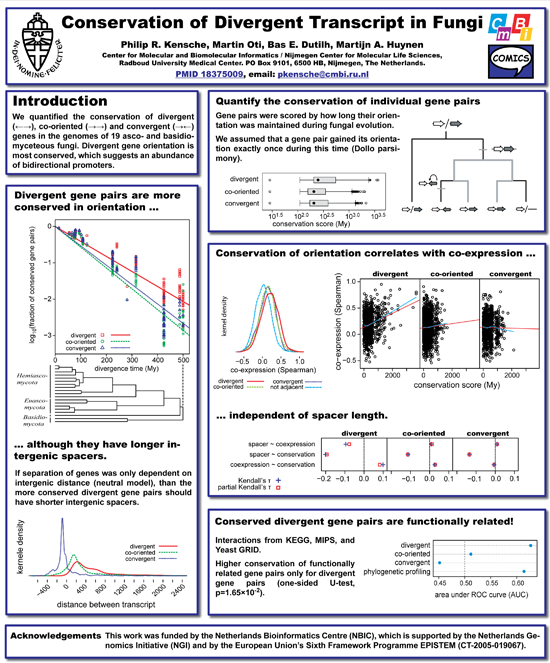
|
|
Philip R. Kensche, Martin Oti, Bas E. Dutilh and Martijn A. Huynen (2007), "Conservation of Gene Orientation in Fungi", poster at ESF-EMBO Symposium "Comparative Genomics of Eukaryotic Microorganisms: Eukaryotic Genome Evolution", Sant Feliu de Guixols, Spain. |
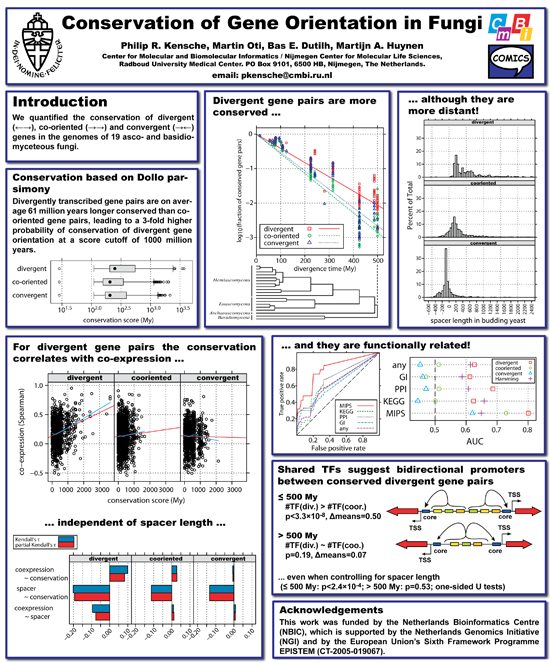
|
Philip R. Kensche, Vera van Noort, Bas E. Dutilh and Martijn A. Huynen (2008), "Practical and theoretical advances in predicting the function of a protein from its phylogenetic distribution", Journal of the Royal Society Interface 5: 151-170. PubMed.
The gap between the amount of genome information released by genome sequencing projects and our knowledge about the proteins' functions is rapidly increasing. To fill this gap, various 'genomic-context' methods have been proposed that exploit sequenced genomes to predict the functions of the encoded proteins. One class of methods, phylogenetic profiling, predicts protein function by correlating the phylogenetic distribution of genes with that of other genes or phenotypic characteristics. The functions of a number of proteins, including ones of medical relevance, have thus been predicted and subsequently confirmed experimentally. Additionally, various approaches to measure the similarity of phylogenetic profiles and to account for the phylogenetic bias in the data have been proposed. We review the successful applications of phylogenetic profiling and analyse the performance of various profile similarity measures with a set of one microsporidial and 25 fungal genomes. In the fungi, phylogenetic profiling yields high-confidence predictions for the highest and only the highest scoring gene pairs illustrating both the power and the limitations of the approach. Both practical examples and theoretical considerations suggest that in order to get a reliable and specific picture of a protein's function, results from phylogenetic profiling have to be combined with other sources of evidence.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Bas E. Dutilh (October 15th 2007), "Extracting the evolutionary signal from genomes", Ph.D. thesis. PDF. Email me to receive a printed copy.
Several methods to analyze aspects of evolution are developed, that depend on the availability of complete genomes. While I consistently find a phylogenetic signal using many approaches, a question that is winning concern is how these evolutionary relationships should be interpreted. Since Darwin's idea about the tree-like structure of evolution, the dogma has been that evolution is mainly a vertical process, but recently, Doolittle pointed out that especially for prokaryotes, a tree may be insufficient to capture the complex evolutionary paths leading to the current-day genomes. While a tree may fall short as a representation of the evolutionary relationships between genomes, I think that describing a species as its entire genome blurs your vision. To characterize a species, I would look at its core, and disregard the noisy genes that obscure its evolutionary history (chapters "Genome trees and the nature of genome evolution", "The Consistent Phylogenetic Signal in Genome Trees Revealed by Reducing the Impact of Noise" and "Assessment of phylogenomic and orthology approaches for phylogenetic inference"). To identify these cores at many different levels throughout the tree of life, I use the hundreds of complete genomes that have become available. In the chapter "Signature genes as a phylogenomic tool", I find signature genes for every clade, and show that these can be used for the taxonomic characterization of a sequenced sample, for example an environmental sample. Another type of data that have become available on a large scale are gene expression data. To be able to compare the functional context of genes in distantly related species, we developed the expression context, that relies on the completeness of the genome sequences and on the availability of genome-wide expression experiments (chapter "A global definition of expression context is conserved between orthologs, but does not correlate with sequence conservation").

Nicole A. Datson, Maarten C. Morsink, Srebrena Atanasova, Victor W. Armstrong, Hans Zischler, Christina Schlumbohm, Bas E. Dutilh, Martijn A. Huynen, Brigitte Waegele, Andreas Ruepp, E. Ronald de Kloet and Eberhard Fuchs (2007), "Development of the first marmoset-specific DNA microarray (EUMAMA): a new genetic tool for large-scale expression profiling in a non-human primate" BMC Genomics 8: 190. PubMed, PDF.
Background The common marmoset monkey (Callithrix jacchus), a small non-endangered New World primate native to eastern Brazil, is becoming increasingly used as a non-human primate model in biomedical research, drug development and safety assessment. In contrast to the growing interest for the marmoset as an animal model, the molecular tools for genetic analysis are extremely limited. Results Here we report the development of the first marmoset-specific oligonucleotide microarray (EUMAMA) containing probe sets targeting 1541 different marmoset transcripts expressed in hippocampus. These 1541 transcripts represent a wide variety of different functional gene classes. Hybridisation of the marmoset microarray with labelled RNA from hippocampus, cortex and a panel of 7 different peripheral tissues resulted in high detection rates of 85% in the neuronal tissues and on average 70% in the non-neuronal tissues. The expression profiles of the 2 neuronal tissues, hippocampus and cortex, were highly similar, as indicated by a correlation coefficient of 0.96. Several transcripts with a tissue-specific pattern of expression were identified. Besides the marmoset microarray we have generated 3215 ESTs derived from marmoset hippocampus, which have been annotated and submitted to GenBank [GenBank: EF214838 - EF215447, EH380242 - EH382846]. Conclusions We have generated the first marmoset-specific DNA microarray and demonstrated its use to characterise large-scale gene expression profiles of hippocampus but also of other neuronal and non-neuronal tissues. In addition, we have generated a large collection of ESTs of marmoset origin, which are now available in the public domain. These new tools will facilitate molecular genetic research into this non-human primate animal model.
Bas E. Dutilh, Vera van Noort, René T.J.M. van der Heijden, Teun Boekhout, Berend Snel and Martijn A. Huynen (2007), "Assessment of phylogenomic and orthology approaches for phylogenetic inference", Bioinformatics 23: 815-824. PubMed, PDF.
Motivation: Phylogenomics integrates the vast amount of phylogenetic information contained in complete genome sequences, and is rapidly becoming the standard for inferring reliable species phylogenies. There are however fundamental differences between the ways in which phylogenomic approaches like gene content, superalignment, superdistance and supertree integrate the phylogenetic information from separate orthologous groups. Furthermore, they all depend on the method by which the orthologous groups are initially determined. Here, we systematically compare these four phylogenomic approaches, in parallel with three approaches for large-scale orthology determination: pairwise orthology, cluster orthology and tree-based orthology. Results: Including various phylogenetic methods, we apply a total of 54 fully automated phylogenomic procedures to the Fungi, the eukaryotic clade with the largest number of sequenced genomes, for which we retrieved a golden standard phylogeny from the literature. Phylogenomic trees based on gene content show, relative to the other methods, a bias in the tree topology that parallels convergence in life style among the species compared, indicating convergence in gene content. Conclusions: Complete genomes are no warrant for good, or even consistent phylogenies. However, the large amounts of data in genomes enable us to carefully select the data most suitable for phylogenomic inference. In terms of performance, the superalignment approach, combined with restrictive orthology, is the most successful in recovering a fungal phylogeny that agrees with current taxonomic views, and allows us to obtain a high resolution phylogeny. We provide solid support for what has grown to be common practice in phylogenomics during its advance in recent years.
Bas E. Dutilh and Martijn A. Huynen (2006), "Superalignment and supertree are the best phylogenomic approaches", talk at International Conference in Phylogenomics, Sainte Adèle, Quebec, Canada. In: Conference Program Phylogenomics Conference, p. 20.
|
Bas E. Dutilh, Vera van Noort, René T.J.M. van der Heijden, Martijn A. Huynen and Berend Snel (2006), "A comprehensive comparison of phylogenomics and orthology methods applied to the Fungi", poster at International Conference in Phylogenomics, Sainte Adèle, Quebec, Canada. In: Conference Program Phylogenomics Conference, p. 40. |

|
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Marc Strous, Eric Pelletier, Sophie Mangenot, Thomas Rattei, Angelika Lehner, Michael W. Taylor, Matthias Horn, Holger Daims, Delphine Bartol-Mavel, Patrick Wincker, Valérie Barbe, Nuria Fonknechten, David Vallenet, Béatrice Segurens, Chantal Schenowitz-Truong, Claudine Médigue, Astrid Collingro, Berend Snel, Bas E. Dutilh, Huub J. M. Op den Camp, Chris van der Drift, Irina Cirpus, Katinka T. van de Pas-Schoonen, Harry R. Harhangi, Laura van Niftrik, Markus Schmid, Jan Keltjens, Jack van de Vossenberg, Boran Kartal, Harald Meier, Dmitrij Frishman, Martijn A. Huynen, Hans-Werner Mewes, Jean Weissenbach, Mike S. M. Jetten, Michael Wagner and Denis Le Paslier (2006), "Deciphering the evolution and metabolism of an anammox bacterium from a community genome", Nature 440: 790-794. PubMed, PDF, F1000 Exceptional.
Anaerobic ammonium oxidation (anammox) has become a main focus in oceanography and wastewater treatment. It is also the nitrogen cycle's major remaining biochemical enigma. Among its features, the occurrence of hydrazine as a free intermediate of catabolism, the biosynthesis of ladderane lipids and the role of cytoplasm differentiation are unique in biology. Here we use environmental genomics the reconstruction of genomic data directly from the environment to assemble the genome of the uncultured anammox bacterium Kuenenia stuttgartiensis from a complex bioreactor community. The genome data illuminate the evolutionary history of the Planctomycetes and allow us to expose the genetic blueprint of the organism's special properties. Most significantly, we identified candidate genes responsible for ladderane biosynthesis and biological hydrazine metabolism, and discovered unexpected metabolic versatility.

Guenola Ricard, Neil R. McEwan, Bas E. Dutilh, Jean-Pierre Jouany, Didier Macheboeuf, Makoto Mitsumori, Freda M. McIntosh, Tadeusz Michalowski, Takafumi Nagamine, Nancy Nelson, Charles J. Newbold, Eli Nsabimana, Akio Takenaka, Nadine A. Thomas, Kazunari Ushida, Johannes H.P. Hackstein and Martijn A. Huynen (2006), "Horizontal Gene Transfer from Bacteria to rumen Ciliates indicates adaptation to their anaerobic carbohydrates rich environment", BMC Genomics 7: 22. PubMed, PDF, F1000 Recommended, ISI.
Background The horizontal transfer of expressed genes from Bacteria into Ciliates which live in close contact with each other in the rumen (the foregut of ruminants) was studied using ciliate Expressed Sequence Tags (ESTs). About 4000 ESTs were sequenced from the two main types of rumen Cilates: Entodiniomorphs (Entodinium simplex, Entodinium caudatum, Eudiplodinium maggii, Metadinium medium, Diploplastron affine, Polyplastron multivesiculatum and Epidinium ecaudatum) and Vestibuliferida, previously called Holotrichs (Isotricha prostoma, Isotricha intestinalis and Dasytricha ruminantium). Results A comparison of the sequences with the completely sequenced genomes of Eukaryotes and Prokaryotes, followed by large scale construction and analysis of phylogenies, identified 148 ciliate genes that specifically cluster with genes from the Bacteria. Of these genes, 34 cluster with genes from the Firmicutes, a phylum of Bacteria that is well represented in the rumen. The phylogenetic clustering with bacterial genes, coupled with the absence of close relatives of these genes in the Ciliate Tetrahymena thermophila, indicates that they have recently been acquired via Horizontal Gene Transfer (HGT). Conclusions Among the HGT candidates, we found an over representation (>75%) of genes involved in metabolism, specifically in the catabolism of complex carbohydrates (>45%), a rich food source in the rumen. We propose that the acquisition of these genes has facilitated the Ciliates' colonization of the rumen and provides evidence for the role of HGT in the adaptation to new niches.
Bas E. Dutilh, Martijn A. Huynen and Berend Snel (2006), "A global definition of expression context is conserved between orthologs, but does not correlate with sequence conservation", BMC Genomics 7: 10 (highly accessed). PubMed, PDF, ISI.
Background The massive scale of microarray derived gene expression data allows for a global view of cellular function. Thus far, comparative studies of gene expression between species have been based on the level of expression of the gene across corresponding tissues, or on the co-expression of the gene with another gene. Results To compare gene expression between distant species on a global scale, we introduce the "expression context". The expression context of a gene is based on the co-expression with all other genes that have unambiguous counterparts in both genomes. Employing this new measure, we show 1) that the expression context is largely conserved between orthologs, and 2) that sequence identity shows little correlation with expression context conservation after gene duplication and speciation. Conclusions This means that the degree of sequence identity has a limited predictive quality for differential expression context conservation between orthologs, and thus presumably also for other facets of gene function.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Berend Snel, Martijn A. Huynen and Bas E. Dutilh (2005), "Genome trees and the nature of genome evolution", Annual Review of Microbiology 59: 191-209. PubMed.
Genome trees are a means to capture the overwhelming amount of phylogenetic information that is present in genomes. Different formalisms have been introduced to reconstruct genome trees on the basis of various aspects of the genome. On the basis of these aspects, we separate genome trees into five classes: (a) alignment-free trees based on statistic properties of the genome, (b) gene content trees based on the presence and absence of genes, (c) trees based on chromosomal gene order, (d) trees based on average sequence similarity, and (e) phylogenomics-based genome trees. Despite their recent development, genome tree methods have already had some impact on the phylogenetic classification of bacterial species. However, their main impact so far has been on our understanding of the nature of genome evolution and the role of horizontal gene transfer therein. An ideal genome tree method should be capable of using all gene families, including those containing paralogs, in a phylogenomics framework capitalizing on existing methods in conventional phylogenetic reconstruction. We expect such sophisticated methods to help us resolve the branching order between the main bacterial phyla.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Bas E. Dutilh, Martijn A. Huynen, William J. Bruno and Berend Snel (2004), "The consistent phylogenetic signal in genome trees revealed by reducing the impact of noise", Journal of Molecular Evolution 58: 527-539. PubMed, PDF, F1000 Must Read.
With the sequencing of complete genomes we have the most complete molecular data for the reconstruction of the phylogeny of life. For example, instead of using the sequence similarity between proteins, as is classically done for the reconstruction of phylogenies, we can now use the number of shared genes between genomes. The goal of this project is to construct phylogenies using these and other types of "complete genome data" in new ways, combining as much of the genomic information as possible. Aside from being interesting in themselves, these genome trees are of extreme importance to detect and filter out various types of phylogenetic bias in genomic data sets, and therewith improve our methods for the prediction of protein interactions.
|
Bas E. Dutilh, Martijn A. Huynen, William J. Bruno and Berend Snel (2003), "The consistent signal in genome trees revealed by reducing the impact of noise", poster at ECCB, Paris, France. Bas E. Dutilh, Martijn A. Huynen, William J. Bruno and Berend Snel (2003), "The consistent signal in genome trees revealed by reducing the impact of noise", poster at 6th Annual Conference on Computational Genomics, Boston, USA. |
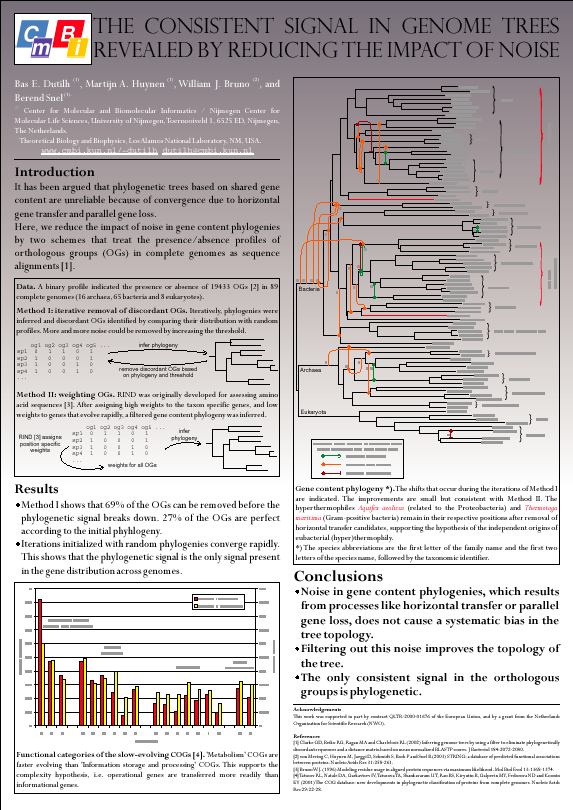
|
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Bas E. Dutilh and Rob J. de Boer (2003), "Decline in excision circles requires homeostatic renewal or homeostatic death of naive T cells", Journal of Theoretical Biology 224: 351-358. PubMed, PDF.
When the TCR is formed in the thymus, fragments of DNA are excised from the T cell progenitor chromosome. These TCR rearrangement excision circles (TRECs) are stable, are not replicated in cell division and are therefore most frequent in naive T cells that have recently left the thymus. During life, the average TREC content of peripheral naive T cells decreases between one and two orders of magnitude in humans. It is generally believed that the age-dependent decrease in the production of naive T cells by the thymus is sufficient to explain the decrease in the TREC content. Here, we demonstrate that this decrease in thymic production is required, but it is not sufficient to explain the TREC data. Only if the decrease in thymic output is compensated by homeostasis can one explain the decrease in the TREC content. The homeostatic response can take two forms: when the total number of naive T cells declines, there could be an increase in the renewal rate or an increase of the average cellular lifespan.
|
Bas E. Dutilh, Rob J. de Boer, Mette D. Hazenberg and Frank M. Miedema (2000), "Decline in excision circles proves homeostasis of naive T-cells", poster at Joint Annual Meeting of Immunology of DGfI and NVvI, Düsseldorf, Germany. |

|
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Bas E. Dutilh (2002), "Pyrobase: integrated database of Pyrococcus genes": www.cmbi.ru.nl/pyrobase.
With the sequencing of complete genomes we can predict the metabolic pathways in a species.
Some of these species are of particular importance, either for medical or for industrial reasons.
The Pyrococci are a genus that belong to the Archaea, one of the three branches in the evolution of cellular life, and the one about which the least is known.
Pyrococcus is a hyperthermophile that lives at about 90°C, and has a large reductive potential.
This makes the organism interesting for the production of certain fine chemicals (alcohols and aldehydes) from carboxyl acids.
The goal of this project is to detect the enzymes that are involved in the carbohydrate metabolism (e.g. glycolysis, citric acid cycle, fatty acid metabolism) of the Pyrococci.
Some of them have already been annotated in the genome, but with the methods that are being developed in our group we have proposed new candidates.
In this partly EU funded project, we work in collaboration with the Bacterial Genetics Group of John van der Oost at Wageningen University, where specific predictions can be tested.
Pyrobase is the integrated database of Pyrococcus genes made as a part of this project. The genomes of Pyrococcus abyssi, Pyrococcus furiosus, Pyrococcus horikoshii, the only three Pyrococcus species with completely sequenced genomes, were screened for Pfam protein families, the genes were assigned to a cluster of orthologous groups of proteins from the COG database, genomically linked orthologous groups were identified with STRING, and buttons are included to directly submit the protein sequence to a STRING genomic context search, and a SMART domain architecture search.
Berend Snel, Toni Gabaldón, Vera van Noort, Bas E. Dutilh and Martijn A. Huynen (2002), "Computational genomics for protein function and pathway prediction", poster at KNCV Symposium "Bioinformatics, the best of both worlds", Wageningen, The Netherlands.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Bas E. Dutilh, Chris E. Dutilh and Willem H.M.M. van Laarhoven (2001), "Initiatives on Sustainable Development in the Food Sector Worldwide", Foundation for Sustainability in the Food Chain (DuVo). PDF.
In 1995 fifteen companies, active in the food chain in The Netherlands have initiated the Foundation for a Sustainable Food Chain (DuVo). The first projects carried out by DuVo were related to the identification of major environmental impacts in the food chain. Subsequently the focus changed to the identification of options for improvement along the production chain and to the development of an infrastructure, which could contain and provide such information. In 1998, DuVo has formulated a new strategy, which is composed of the following elements:
- A dialogue with relevant stakeholders, aimed at establishing a common definition for the concept 'sustainable food chain'. In that process, measurable criteria can be developed to manage and monitor an improvement process;
- Development of knowledge, aimed at providing factual information which can improve the content of the dialogue;
- Open exchange of knowledge to enable as many parties as possible to share the insights which have been acquired.
| 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001 | 1999 |
Bas E. Dutilh and Paulien Hogeweg (1999), "Gene Networks from Microarray Data", report Binf.1999.11.01, Bioinformatics, Utrecht University: www.cmbi.ru.nl/~dutilh/genenets. Thesis site, PDF.
Since the development of the microarray technique in 1995, there has been an enormous increase in gene expression data from several organisms. Based on the view of gene systems as a logical network of nodes that influence each other's expression levels, scientists dream of being able to reconstruct the precise gene interaction network from the expression data obtained with this large scale arraying technique. Computer science shows that inference of a logical regulatory network is possible solely from sets of expression data, and mathematicians are working on the question how much data is at least necessary for reverse engineering.
Meanwhile, experimental biologists are experiencing problems in the field. The number of experiments that are necessary before attempting network reconstruction is a lot more than is generally possible in "wet" laboratories, so data compression algorithms are applied to reduce the number of nodes considered. This is however an extremely coarse representation of the intricate interconnections that exist between single genes. The resulting network of only a handful of nodes is therefore usually only sufficient to describe the experiments performed, while any possible predicting properties are absent.
In this literature thesis, I attempt to give an update on the state of the art in computerised network reconstruction techniques, and explicitly relate this to actual biological gene networks. I will go into the model formalisms used to describe genetic networks, and explain their specific advantages and disadvantages. Also, a separate chapter will be dedicated to several experimental results obtained in the research of genetic networks, and finally, a short discussion and some hypothesising is added.
Bas E. Dutilh and Paulien Hogeweg (1999), "Evolution of viral strain structure through host immune response", talk at TMBM'99, Amsterdam, The Netherlands. In: abstracts of TMBM'99, Amsterdam: 160-161. PDF.
Recently, it was shown that host immune responses can form a strong selective pressure on the antigenic strain structure of pathogen populations. In an ODE model described by Gupta et al. [1], the evolutionary dynamics of infective agents (each viral strain is defined as a specific combination of several alleles at a number of epitopes) can lead to discrete strain structures, called discordant sets. Discordant sets consist of viruses which have no antigens in common, and together fill up the complete antigen space with their genotypes. These sets of infective agents inhibit the spreading of related pathogens in a host population, by making the hosts resistant to all antigens in the world.
We further examine potential emergent strain structures due to host immune responses, in a spatially explicit model including a population of immunologically reactive hosts. The hosts, which are individually implemented in a cellular automata machine, can each carry their own virus, and are each resistant to a specific combination of antigens. Thus, we are able to study many different assumptions on immune systems in one simple model. In the present study, for example, a host that is infected with several viruses, can collect resistances against all their antigens. Upon encounter of a newly attacking infection, it will oppose an immunity that is proportional to the amount of the infectious agent's antigens it has gathered immunity to.
Surprisingly, we discovered that spatial pattern formation in the cellular automata machine is necessary for generation of discordant strain structure such as found in the ODE model of Gupta et al. [1]. In the case of a mean field approximation (randomly reshuffling the cellular automata every timestep), agglomerative clustering techniques reveal strain structure in larger sets of viruses. There is a clear selection for minimization of the encountered immunities, and for each virus, this is optimized in a set with symmetric and minimum amounts of antigenic overlaps. Though these conditions are satisfied in any collection of discordant sets (including the complete viral population) the cofluctuating sets never contain discordants. The observed strain structures allow for larger virus populations, causing more infections during the lifetime of a host than an equal number of viruses organized in discordant sets would.
We conclude that host immune response can structure viral populations into discrete sets, which are not invadable by new mutants. Moreover, we see that spatial pattern formation, which leads to discordant sets of viruses, protects the hosts and reduces the numbers of viruses that survive.
[1] S. Gupta, N. Ferguson & R. Anderson: "Chaos, Persistance, and Evolution of Strain Structure in Antigenically Diverse Infectious Agents", Science, 280: 912-915, 1998.